 2025, Vol. 45
2025, Vol. 45文章信息
- 罗钰燕, 王宇姝, 乐晶, 盛海彦, 余玲玲
- LUO Yuyan, WANG Yushu, LE Jing, SHENG Haiyan, YU Lingling
- 青藏高原盐湖/超盐湖表层沉积物原核生物群落分布格局及其形成机制
- Distribution pattern and formation mechanism of prokaryotic community in surface sediments of saline/hyper-saline lakes on the Qinghai-Tibet Plateau
- 生态学报. 2025, 45(10): 5025-5042
- Acta Ecologica Sinica. 2025, 45(10): 5025-5042
- http://dx.doi.org/10.20103/j.stxb.202408071859
-
文章历史
- 收稿日期: 2024-08-07
- 网络出版日期: 2025-03-18






2. 华南农业大学资源环境学院生态系, 广州 510642;
3. 青海大学农牧学院, 西宁 810016
2. Department of Ecology, College of Natural Resources and Environment, South China Agricultural University, Guangzhou 510642, China;
3. College of Agriculture and Animal Husbandry, Qinghai University, Xining 810016, China
高寒盐湖的表层沉积物中蕴藏着丰富的原核微生物群落, 它们是湖底沉积物食物网的重要组成部分, 也是驱动生物地球化学循环的主要参与者[1]。在高寒盐湖生态系统中, 原核微生物在土壤形成、有机质代谢、养分转化以及能量流动代谢中都具有不可替代的作用, 这些原核微生物细胞结构简单, 但对高寒湖泊的生态环境具有极强的耐受和适应能力[2]。位于青藏高原盐湖中的原核微生物因其地理上的特殊位置, 使得它们对环境变化的响应表现出高度的敏感性和快速性[3]。因此, 原核微生物群落结构的动态变化能够作为反映生态系统的功能和稳定性一个敏感指标。这一特性对于深入理解高寒盐湖生态系统中原核微生物群落分布格局, 以及评估其对生态系统服务和生物多样性保护的潜在影响具有重要的科学意义。
青海湖和茶卡湖是青藏高原上具有代表性的盐湖。茶卡湖属于超盐湖泊, 总盐度趋于饱和。在这种极端环境下, 细菌生长代谢受到限制, 生物多样性偏低[4]。相比之下, 青海湖的盐度范围适中, 细菌群落的多样性较高[5]。近年来, 越来越多的研究集中在青藏高原盐湖中原核微生物群落变化及其环境调控因素上[5—7], 这些研究发现盐度是影响原核微生物群落丰度与多样性的最重要因素。然而已有研究通常集中在单一生物群体, 并不能在群落水平上全面理解细菌和古菌在环境变化下生态功能的统一性和差异, 对原核微生物在盐胁迫下的响应及群落间相互作用的理解仍然有限。
有研究表明, 利用β-多样性分解分析可以有效地发掘微生物群落分布特征和形成的机制[8]。β-多样性分解是指不同群落间物种组成的差异由物种的周转组分和嵌套组分这两种过程决定[9]。物种的周转和嵌套组分对总β-多样性的相对重要性因地理、空间、生境类型和物种类群等而异。许多研究发现总β-多样性由物种周转过程主导[10—11], 表明环境因素和扩散限制在很大程度上影响了β-多样性的分布模式。更确切地说, 扩散能力强的物种能更容易跨越地理障碍, 形成较低的总β-多样性和较高的周转组分, 而扩散能力较弱的物种更容易受到环境隔离的影响, 从而形成较高的总β-多样性和嵌套组分[8]。目前分析周转和嵌套组分对高原湖泊中原核微生物中总β-多样性相对贡献的研究还鲜有报道。除此之外, 分子生态网络分析已被广泛用于探索高原湖泊沉积生态系统中微生物的相互作用[6, 12]。微生物群落并不是作为单个种群单独存在于湖泊生态系统中, 不同群落之间经常共存并产生的相互作用并形成复杂的网络, 这些相互作用对微生物群落的形成至关重要[13]。分子生态网络分析技术能够更进一步地探究微生物种间的相互作用, 评估微生物群落结构变化引起的生态系统功能演变以及生态系统的稳定性[14]。
本文选取青海湖和茶卡湖表层沉积物的原核微生物为研究对象, 采用β-多样性分解、偏最小二乘法结构方程模型(partial least square-structural equation modeling, PLS-SEM)和生态网络分析等方法研究在不同环境下, 周转组分与嵌套组分相对贡献的变化, 并分析青海湖和茶卡湖表层沉积物中原核微生物群落之间的相互作用。本研究旨在深入探讨高原盐湖中原核微生物群落的多样性、生态功能以及在极端环境下的生存策略, 揭示高寒湖泊中原核微生物群落的特征、生物地理分布及其背后的驱动机制。通过对现象及其本质的深入分析, 本研究为高寒湖泊生态系统中原核微生物群落的结构和分布模式提供了实证数据和理论支持。这些发现对于微生物生物地理学的发展、微生物多样性资源的保护具有重要理论意义。
1 材料与方法 1.1 采样地点与方法青藏高原的湖泊大多是碱性的, 根据湖泊的盐度水平, 将其分为低盐湖(< 1 g/L)、中度盐湖(1—35 g/L)和高盐湖(> 35 g/L)[15]。青海湖(36°32′—37°15′ N、99°36′—100°16′ E)位于青藏高原东北部, 平均海拔3196 m。青海湖是一个封闭性的咸水湖, 湖面面积4300 km2, 平均盐度为15.5 g/L[16]。茶卡湖(36°18′—36°45′ N、99°02′—99°12′ E)位于柴达木盆地, 海拔3214 m, 湖面面积为105 km2。茶卡湖是一个天然结晶高盐湖, 由于该地区蒸发量大, 降水量少, 盐度为299.3—350.7 g/L[17], 处于盐饱和状态。本研究在2015年5月分别在青海湖东北5个采样点(QSe1、QSe2、QSe3、QSe4、QSe5)和茶卡湖3个采样点(CSe1、CSe2、CSe3)共8个采样点采集了表层沉积物样品, 每个采样点3个重复, 共有24个样品。QSe4和QSe5采样点位于青海湖北岸, CSe2采样点位于采盐河道, CSe3采样点位于茶卡湖码头附近, 每个表层沉积物采样点至少相距2 km, 各采样点的地理位置分布如图 1所示。采用抓斗式采泥器在青海湖、茶卡盐湖和青海湖北岸底抓取水深2至3 m处的沉积物样品, 表层沉积物采样深度为表层0—5 cm。用无菌刮刀将表层沉积物样本放入50 mL的无菌试管中。采样后立即放入便携式冰盒, 一部分于-20℃冷冻保存并于2015年7月送至上海美吉生物医药科技有限公司进行高通量测序, 另一部分于4℃条件下保存用于测定湖底表层沉积物的理化性质。

|
| 图 1 采样点位置图 Fig. 1 Sampling sites |
在取样地点使用电导率仪(多参数探头, Orion Star A325, Thermo Fisher, USA)测量沉积物电导率(EC)[18], 并根据《湖泊沉积物调查规范》[19]和《土壤农化分析》[20]测定以下沉积物理化性质:
(1) 用玻璃电极法(Eutech pH玻璃电极, Thermo Fisher, USA)测定沉积物pH(土∶水=1 ∶ 2.5)。
(2) 用重铬酸钾-外加热法测定沉积物有机碳(SOC)含量。具体操作为称取0.1 g风干沉积物样置于硬质试管中, 加入10 mL重铬酸钾-硫酸标准溶液(0.4 mol/L), 同时做3组空白试剂(不加沉积物样)。将盛有沉积物样的硬质试管插入油浴锅内的铁丝笼架中加热, 控制油浴锅内温度在170—180℃, 使溶液保持沸腾5 min后取出冷却。随后将试管内容物全部洗入250 mL三角瓶中, 向待测液中滴加3滴10 g/L邻啡罗啉指示剂, 用硫酸亚铁标准溶液(0.2 mol/L)滴定至溶液变棕红色。最后根据以下公式计算出沉积物的有机碳含量: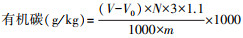
(3) 用半微量凯氏定氮法测定沉积物总氮(TN)。具体操作为称取1 g风干沉积物样品放入凯氏烧瓶反应管中, 加入3.6 g混合催化剂(由3 g硫酸钾, 0.5 g硫酸铜和0.1 g硒粉磨碎混匀制成), 并与试样混合均匀, 同时做3组空白试剂(不加沉积物样)。随后加入12 mL浓硫酸和2粒玻璃珠, 开启加热系统进行消解, 待内容物全部焦化状后滴加1 mL过氧化氢并保持瓶内液体微沸, 直至消化液呈灰白色浑浊状, 继续加热至少1 h。溶液冷却后加入60 mL蒸馏水并转入100 mL容量瓶中作为试样分解液。移取10 mL分解液注入蒸馏装置的反应管中, 加入10 mL氢氧化钠标准溶液(0.1 mol/L)后蒸馏5 min, 再用盐酸标准溶液(0.1 mol/L)滴定蒸馏后的分解液直至溶液变红。最后根据以下公式计算出沉积物的总氮含量: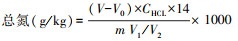
(4) 用钼锑比色法测定总磷(TP)。具体操作为称取1 g风干沉积物样放入消煮管中, 滴入3滴蒸馏水和8 mL浓硫酸, 摇匀后放置8 h, 同时做3组空白试剂(不加沉积物样)。随后加入10滴高氯酸溶液后再放置消解炉中加热至300℃, 消解约1 h至溶液变白, 冷却至常温, 再用蒸馏水将消煮液洗入100 mL的容量瓶中并定容。用2层无磷滤纸过滤后取出3 mL滤液至50 mL离心管, 再用蒸馏水定容至30 mL。然后加入2滴二硝基酚指示剂(0.2%)和6滴氢氧化钠溶液(2 mol/L)调节pH至溶液刚呈微黄色, 再加入5 mL钼锑抗显色剂, 摇匀后用水定容至50 mL。室温放置10 min后同磷标准系列溶液一起用分光光度计(Cary 60 UV-Vis, Agilent, USA)在波长700 nm比色, 最后根据以下公式计算出沉积物的总磷含量: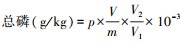
(5) 用火焰光度法测定总钾(TK)。具体操作为称取0.1 g风干沉积物后放入坩埚中, 加入3 mL硝酸和0.5 mL高氯酸, 同时做3组空白试剂(不加沉积物样)。置于电热板上加热至出现大量白烟, 样品成糊状时取下冷却。然后加入5 mL氢氟酸, 0.5 mL高氯酸, 随后在电热板上加热, 温度控制在200℃。继续加热至不再冒白烟时, 加入10 mL盐酸溶液(3 mol/L), 再继续加热至残渣溶解。取下冷却后加入2 mL硼酸溶液(2%), 然后用蒸馏水将溶液转入100 mL容量瓶中, 定容混匀, 作为分解液, 并同钾标准系列溶液一起用火焰光度计(Sherwood M410, UK)测定。最后根据以下公式计算出沉积物的总钾含量: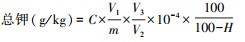
(6) 用水蒸气蒸馏法测定有效氮(AN)。具体操作为称取3 g风干沉积物后放入消化管内, 加入1 g锌-硫酸亚铁还原剂(50 g硫酸亚铁和10 g锌粉混匀制成), 20 mL蒸馏水和1 mL液状石蜡, 同时做3组空白试剂(不加沉积物样)。在全自动定氮仪(FOSS Kjeltec 8400, Denmark)上加入10 mL氢氧化钠和30 mL硼酸指示剂(10 g/L), 蒸馏终点体积为150 mL, 并在滴定剂桶中加入已标定的盐酸溶液(0.1 mol/L)后进行分析。最后根据以下公式计算出沉积物的有效氮含量: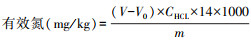
(7) 用比色法测定有效磷(Olsen_P)。具体操作为称取2.5 g风干沉积物样置于200 mL容量瓶中, 加入50 mL碳酸氢钠溶液(0.5 mol/L), 同时按相同步骤制备空白溶液(不加沉积物样)。吸取10 mL试样溶液于50 mL容量瓶并加入5 mL钼锑抗显色剂, 再加入10 mL蒸馏水, 摇匀后室温静置30 min。然后同磷标准系列溶液一起在分光光度计(Cary 60 UV-Vis, Agilent, USA)在波长660 nm进行比色。最后根据以下公式计算出沉积物的有效磷含量: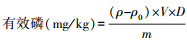
(8) 用原子吸收法测定有效钾(AK)。具体操作为称取2.5 g风干沉积物于250 mL容量瓶中, 加入50 mL浓硝酸浸提剂(2 mol/L), 同时按相同步骤制备空白溶液(不加沉积物样)。在室温下将容量瓶放置在震荡器中震荡30 min, 静止30 min后用定量滤纸过滤, 滤液转移到50 mL离心管中。分别吸取2 mL滤液到10 mL离心管中, 各加入1 mL氯化钠(10 g/L)和7 mL蒸馏水后摇匀, 同钾标准系列溶液一起在原子吸收光谱仪(iCE 3500 AAS, Thermo Fisher, USA)测定。最后根据以下公式计算出沉积物的有效钾含量: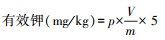
沉积物总DNA采用PowerSoil DNA isolation kit(MoBio, USA)试剂盒提取。提取方法按试剂盒说明书进行。采用Nanodrop 2000(Thermo, UAS)测定沉积物总DNA浓度及纯度。纯化、质控后, 在Illumina MiSeq平台进行Paired-end 250测序(美吉生物, http://www.majorbio.com)。测序引物选取细菌16S rRNA V4-V5区域的515F(3′-GTGCCAGCMGCCGCGG-5′)/907R(3′-CCGTCAATTCMTTTRAGTTT-5′)和古菌16S rRNA V3—V5区域的Arch344F(5′-ACGGGGYGCAGCAGGCGCGA-3′)/Arch915R(5′-GTGCTCCCCCGCCAATTCCT-3′)。序列数据保存在国家微生物学数据中心(https://nmdc.cn/resource/genomics/sample/detail/), 项目编号NMDC20015769。测序结果使用Flash软件和Trimmomatic软件拼接并去除低质量序列, 根据序列两端的序列结构和引物序列得出有效序列, 校正序列方向。使用UPARSE version 7.1(http://drive5.com/uparse/)软件将相似度阈值97%以上的序列归为一个operation taxonomic unit(OTU), 生成OTU表格, 采用RDP classifier贝叶斯算法对OTU代表序列进一步分类学分析, 统计不同样本的群落组成。
1.4 偏最小二乘法结构方程模型偏最小二乘法结构方程模型可以直观地看出表层沉积物环境因子与原核微生物群落多样性之间的关系, 并进一步计算各关系之间的相关系数。根据相关性和回归分析, 选择影响因子总氮、总磷、有效氮、有效钾作为模型假设。初始模型假设电导率通过影响沉积物营养物质和pH间接影响原核微生物的α-和β-多样性。模型估计采用偏最小二乘法对模型进行了修订和检验, 删除不重要和不相关的途径。采用SmartPLS v4.0建立并分析偏最小二乘法结构方程模型, 计算R方值和拟合优度(GoF)以评估模型的拟合度和预测准确性[21], 并用Bootstrapping算法(bootstrapping=3000)以评估路径系数的显著性。
1.5 网络构建与分析根据随机矩阵理论(RMT)构建了系统发育分子生态网络(pMEN), 分析8个采样点表层沉积物细菌和古菌群落之间的相互作用。主要步骤如下:在pMENA网站(http://ieg4.rccc.ou.edu/MENA/login.cgi)[22]上传微生物OTU水平丰度表, 根据样品数量设置合适的主要参数。缺失数据(Missing data)参数选择在具有配对有效值的空白处填写0.01(Only fill with 0.01 in blanks with paired valid values), 转换(Transformation)选项选择对数转换(Logarithm transformation), 相关矩阵(Similarity matrix)采用皮尔森相关系数(Pearson Correlation Coefficient)计算不同OTU间的两两相似性。运行后可以得到基于随机矩阵原理(RMT)的合适阈值, 构建网络采用的相似性阈值选择0.95。通过对数标准化处理建立OTU表, 并通过计算任意两个OTU的皮尔逊相关性生成相关矩阵, 将相关矩阵转换为相似矩阵, 再将相似矩阵转换为邻接矩阵, 计算OTU节点间的连接强度, 然后根据相似性矩阵表征网络特性。最后将在pMENA网站运行分析后生成的网络文件、节点属性文件和边属性文件导入Cytoscape 3.8.0软件[23]进行可视化处理可得到完整网络。
1.6 统计分析采用SPSS 25.0软件, 通过单因素方差分析比较不同样本之间沉积物的理化性质。采用典型对应分析法(CCA)研究原核微生物群落的相对丰度与沉积物理化性质之间的关系。根据Bray-Curtis距离进行相似性分析(ANOSIM), 以检验不同采样点细菌和古菌群落组成的差异。根据Jaccard指数, 将两两群落之间的总β-多样性分解为两个组成部分, 即周转和嵌套。使用R平台上的“beta part ”包[24]计算总β-多样性中周转和嵌套数值。采用SPSS 25.0软件对总β-多样性、周转和嵌套数据进行单因素方差分析, 用Duncan进行差异显著性分析。
2 结果与分析 2.1 青海湖和茶卡盐湖表层沉积物的化学性质和盐度从整体上看, 在两个盐湖的8个取样点间表层沉积物的化学性质及养分状况表现出明显的差异(表 1)。在青海湖的5个样点中, 有效氮和有效磷的含量出现了明显的分化, 其中, 在QSe1—QSe3取样点的有效氮和有效磷的含量明显高于QSe4和QSe5两个样点。在茶卡湖中, 有效氮含量依次为CSe1>CSe2>CSe3, 有效磷含量依次为CSe3>CSe2>CSe1。青海湖的pH值和总氮含量明显高于茶卡湖, 其中QSe5的pH值和QSe3的TN含量最高。而茶卡湖的有效钾和电导率明显高于青海湖, 且CSe1的有效钾和电导率最高。八个采样点的电导率范围在1312—35220 μS/cm之间, 其中, CSe1的电导率最高(35000 μS/cm以上), 其次是CSe2和CSe3, 介于28000—33000 μS/cm之间。电导率最低的是QSe4和QSe5, 平均值分别为1312.67和8500 μS/cm。从表 1可知茶卡湖(CSe1-CSe3)盐度最高, 基本上是盐饱和。青海湖(QSe1—QSe3)盐度次之, 青海湖北岸(QSe4—QSe5)盐度最低。八个取样点的有机碳、总钾和总磷均无明显差异。
| 采样点 Sample site |
经纬度 Coordinate |
温度 Temperature/℃ |
pH | 有机碳 SOC/(g/kg) |
总氮 TN(g/kg) |
碳氮比 C/N |
总钾 TK/(g/kg) |
总磷 TP/(g/kg) |
有效磷 Olsen_P/(mg/kg) |
有效氮 AN/(mg/kg) |
有效钾 AK/(mg/kg) |
电导率 EC/(μS/cm) |
| QSe1 | 36°36′10″N, 100°31′53″E | 6.0 | 9.02±0.01b | 33.24±0.23d | 3.3±0.04c | 10.08±0.19f | 12.28±0.02e | 1.4±0.01b | 790.57±3.63b | 216.69±0.81b | 680.00±0.00fg | 12226.67±14.53f |
| QSe2 | 36°39′16″N, 100°30′15″E | 7.2 | 8.5±0.02d | 27.71±0.36e | 2.6±0.01e | 10.65±0.18ef | 16.00±0.16b | 1.25±0.02c | 582.91±0.11c | 206.07±0.41c | 653.33±3.33g | 15248.33±9.28d |
| QSe3 | 36°35′10″N, 100°29′49″E | 8.9 | 8.59±0.01c | 41.47±0.07b | 3.49±0.02a | 11.88±0.04de | 14.39±0.02c | 1.5±0.01a | 1488.01±7.29a | 297.58±0.41a | 730.00±0.00f | 13145.00±25.98e |
| QSe4 | 36°50′48″N, 100°40′06″E | 8.0 | 9.02±0.01b | 44.98±0.43a | 3.38±0.03b | 13.31±0.02d | 4.9±0.03g | 0.52±0.01g | 390.67±1.89d | 25.33±0.00g | 923.33±3.33d | 1312.67±0.33h |
| QSe5 | 36°50′10″N, 100°40′16″E | 8.5 | 9.15±0.00a | 38.15±0.21c | 3.21±0.02d | 11.88±0.09de | 5.24±0.01f | 0.55±0.00f | 290.15±1.86e | 19.4±0.21h | 850.00±0.00e | 8500.00±11.55g |
| CSe1 | 36°44′30″N, 99°05′18″E | 9.3 | 8.13±0.01f | 18.83±0.16g | 0.83±0.00f | 22.69±0.20c | 19.00±0.05a | 1.02±0.01d | 220.34±2.10f | 57.69±1.41d | 2900.00±0.00a | 35280.00±34.64a |
| CSe2 | 36°41′51″N, 99°06′43″E | 9.6 | 7.94±0.01g | 45.11±0.07a | 0.45±0.00g | 99.53±0.87a | 5.01±0.12fg | 0.36±0.01h | 282.64±7.42e | 33.77±0.81e | 1533.33±33.33c | 33486.67±29.06b |
| CSe3 | 36°45′20″N, 99°05′28″E | 9.0 | 8.23±0.01e | 26.36±0.06f | 0.51±0.01g | 51.39±0.98b | 12.64±0.01d | 0.76±0.01e | 381.81±20.21d | 30.96±0.81f | 2600.00±57.74b | 29226.67±701.77c |
| QSe1—5:青海湖沉积物1—5 Lake Qinghai sediment1—5;CSe1—3:茶卡湖沉积物1—3 Lake Chaka sediment1—3;Temp:温度Temperature; SOC:有机碳Soil organic carbon; TN:总氮Total nitrogen; C/N:碳氮比Carbon-nitrogen ratio; TK:总钾Total potassium; TP:总磷Total phosphorus; Olsen_P:有效磷Olsen phosphorus; AN:有效氮Available nitrogen; AK:有效钾Available potassium; EC:电导率Electrical conductivity; 数值为平均值;同列数字后不同小写字母表示利用Duncan检验有显著性差异(P < 0.05) | ||||||||||||
采用Illumina MiSeq(PE250)对两种类型湖泊的表层沉积物样本进行测序分析, 得到654116条细菌原始序列和668428条古菌原始序列, 其中包含细菌1326个OTU和古菌1745个OTU。稀释曲线图(图 2)可以看出随着细菌和古菌测序数量的升高, 稀释曲线的Sobs指数虽然还存在增加的趋势, 但是趋近于平缓, 说明测序数据量达到了可分析水平。

|
| 图 2 稀释曲线图 Fig. 2 Rarefaction curve |
α-多样性表征了微生物群落的丰富度和多样性, 本研究中采用Chao和Shannon指数对细菌和古菌群落的丰富度和多样性进行评价(图 3)。青海湖QSe1—QSe5细菌群落的Chao和Shannon指数明显高于茶卡盐湖CSe2-CSe3的群落指数。在QSe1—QSe5样点中, 各样点间的细菌群落丰富度和α-多样性无明显差异, 而在茶卡盐湖中, 样点CSe1细菌群落的丰富度和α-多样性显著高于样点CSe2和CSe3。青海湖的细菌丰富度和α-多样性最高且与茶卡湖差异显著。与细菌群落相比, 古菌群落的丰富度和α-多样性显示出不同的差异。QSe1—QSe5古菌群落的Chao指数普遍高于茶卡湖CSe1-CSe3。QSe1和QSe3古菌群落的Chao指数最高且与其他样本差异显著。在所有样本中, QSe1古菌群落的Shannon指数最高, QSe4和QSe5古菌群落的Shannon指数无显著差异。CSe2古菌群落的Shannon指数最低且与其他样本差异显著。总体而言, 与茶卡湖相比, 青海湖表层沉积物的细菌和古菌群落都表现出更高的丰富度和多样性, 尤其细菌群落的差异更为明显。

|
| 图 3 不同湖泊沉积物原核微生物ɑ-多样性 Fig. 3 ɑ-diversity of prokaryotic community in different lake sediments QSe1—5:青海湖沉积物1—5;CSe1—3:茶卡湖沉积物1—3;柱子上方不同小写字母表示利用Duncan检验有显著性差异(P < 0.05) |
对8个样本表层沉积物的原核微生物群落相对丰度在门水平进行分析。所有样本中门水平的细菌类群主要为Proteobacteria(34.10%)、Bacteroidetes(25.10%)和unclassified_k_norank(11.43%), 其中unclassified_k_norank主要分布在CSe1-CSe3中, 尤其是CSe3。Acetothermia(7.03%)是茶卡盐湖独有的优势类群。所有样本中门水平的古菌群落主要有Euryarchaeota(64.75%)和Woesearchaeota__DHVEG-6(27.25%)。所有样本中都广泛分布Euryarchaeota, 与青海湖QSe1—QSe5(44.43%)相比, 茶卡湖CSe1-CSe3(87.79%)的Euryarchaeota更为丰富。Woesearchaeota_dhveg6是QSe1—QSe5的优势类群之一(38.16%), 在QSe1尤其丰富(77.65%)。总体而言, 青海湖和茶卡湖的细菌和古菌群落组成和丰度存在明显差异, 茶卡湖存在更多的未知原核微生物类群和独特物种。
2.3 原核生物组成及β-多样性组分分析基于Bray-Curtis距离的PCoA分析显示, 青海湖与茶卡湖之间的细菌和古菌群落组成存在明显的分离(图 4)。其中PC1和PC2分别解释了47.22%和17.43% 的细菌群落组成变化, PC1和PC2分别解释了36.86%和21.16% 的古菌群落组成变化。用ANOSIM相似性分析青海湖和茶卡湖表层沉积物的细菌(R2=0.8806, P=0.001)和古菌(R2=0.836, P=0.001)群落组成分布存在显著差异, 进一步检验了青海湖与茶卡湖采样点间细菌和古菌组间差异大于组内差异。

|
| 图 4 不同湖泊沉积物原核微生物的主坐标分析 Fig. 4 The principal co-ordinates analysis(PCoA)of prokaryotic community in different lake sediments |
总β-多样性的驱动过程分为两个部分:周转和嵌套。CSe1细菌群落的总β-多样性与嵌套显著高于其他样本, 其他样本之间无显著差异。在周转部分, CSe1与CSe2和CSe3的细菌群落无显著差异。古菌群落的总β-多样性与细菌群落的变化一致, 古菌群落的周转和嵌套部分相比细菌群落表现出不同的差异。在嵌套部分中, 除CSe2, CSe1显著高于其他样本。CSe3古菌群落的周转部分显著低于CSe1、QSe2和QSe4, 其他样本之间没有显著差异(图 5)。青海湖和茶卡湖表层沉积物中原核微生物群落的总β-多样性主要受嵌套的影响。

|
| 图 5 不同湖泊沉积物原核微生物群落betapart分析 Fig. 5 Betapart analysis of the prokaryotic community in different lake sediments |
基于CCA分析来解释环境因子对不同湖泊表层沉积物中原核微生物群落的影响(图 6)。环境因子对青海湖和茶卡湖表层沉积物细菌群落的总解释量为40.81%, 前两个轴分别解释了26.44%和14.37%的细菌群落变异。环境因子对青海湖和茶卡湖表层沉积物古菌群落的总解释量为38.98%, 前两个轴分别解释了22.46%和16.52%。QSe1—QSe5的细菌和古菌群落主要受到pH、总氮、总磷、有效磷和有效氮的影响, CSe1—CSe3的细菌和古菌群落主要受到有效钾、电导率和碳氮比的影响。

|
| 图 6 不同湖泊沉积物原核微生物群落与环境因子CCA分析 Fig. 6 Canonical Correspondence Analysis(CCA)of prokaryotic community in different lake sediments |
PLS-SEM分析量化环境因子对原核微生物群落多样性和结构的影响(图 7)。电导率是影响青海湖表层沉积物中营养物质(R2=0.867)和pH(R2=0.436)的最大影响因子, 并进一步影响原核微生物群落的α-和β-多样性。青海湖表层沉积物中营养物质对古菌α-多样性有直接的正向影响, 而对细菌和古菌群落的β-多样性有负向影响。pH值直接对青海湖的古菌α-多样性、细菌和古菌的β-多样性产生正向影响。在茶卡湖中, 电导率和pH是影响表层沉积物中的营养物质的最大因素(R2=0.880), 电导率对pH值没有影响。茶卡湖表层沉积物中营养物质对细菌α-和β-多样性都有直接的正向影响。pH值对茶卡湖表层沉积物中的营养物质和古菌α-多样性有显著正向影响, 对古菌β-多样性有显著负面影响。青海湖(GoF=0.6753)和茶卡湖(GoF=0.7382)中原核微生物群落的偏最小二乘法结构方程模型的拟合优度指标均大于0.6, 表明模型估计结果可靠。

|
| 图 7 不同湖泊沉积物原核微生物群落PLS-SEMs分析 Fig. 7 Partial least squares structural equation models(PLS-SEMs)of prokaryotic community in different lake sediments TN:总氮;TP:总磷;AN:有效氮;AK:有效钾;GoF:拟合优度;路径上的数字表示标准化路径系数;*表示路径系数的显著性水平,*P < 0.05, * *P < 0.01, * * * P < 0.001 |
本研究基于原核微生物的OTU数据对青海湖和茶卡湖的表层沉积物进行原核微生物网络分析。细菌网络中的节点主要由门水平上的Proteobacteria、Bacteroidetes、Chloroflexi、Firmicutes、Latescibacteria、Lentisphaerae、Gemmatimonadetes、Spirochaetae等组成。在古菌网络中, 主要的节点包括Euryarchaeota、Thaumarchaeota、Miscellaneous_Euryarchaeotic_Group_MEG、Woesearchaeota_DHVEG-6、Lokiarchaeota、Diapherotrites等(图 8)。青海湖和茶卡湖细菌网络中正负连接的比例主要表明了不同类群之间的相互作用关系。在细菌网络中, QSe2、QSe3、QSe5和CSe3观测到了比较多的正向相互作用, QSe1、QSe4、CSe1和CSe2的正负连接比例达到了平衡。古菌网络显示出与细菌网络不同的模式。正相关在QSe3、QSe5和CSe3的古菌网络中占主导地位, 负相关在QSe2和CSe1上占主导, 正负连接的比例在QSe1、QSe4和CSe2达到平衡, 而CSe3只表现出正的相互作用。

|
| 图 8 不同湖泊沉积物原核微生物菌群间的网络相互作用 Fig. 8 Ecological networks of prokaryotic community in different lake sediments |
原核微生物共现网络在青海湖和茶卡湖之间表现出显著差异。青海湖原核微生物网络的节点数、连接数和平均连通度(网络中每个节点的平均连接数)均较高, 这表明青海湖原核微生物网络的复杂性有所增加。与茶卡湖相比, 青海湖的平均路径长度更短, 平均聚集系数和网络密度更低(表 2)。此外, 具有最大特征向量中心性的节点OTU2614(Hydrogenophaga)、OTU1249(未知)、OTU919(未知)和OTU58(Halapricum)分别是细菌和古菌网络中的关键类群。
| 特征参数 Characteristic parameters |
青海湖Lake Qinghai(0.95) | 茶卡湖Lake Chaka(0.95) | |||
| 细菌 Bacteria | 菌 Archaea | 细菌 Bacteria | 古菌 Archaea | ||
| 节点数 Total nodes | 346 | 138 | 206 | 125 | |
| 连接数 Total links | 1098 | 355 | 906 | 400 | |
| 平均连通度 Average degree | 6.347 | 5.145 | 8.796 | 6.4 | |
| 平均聚集系数 Average clustering coefficient | 0.382 | 0.416 | 0.512 | 0.504 | |
| 平均路径长度 Average path distance | 4.411 | 2.646 | 5.923 | 4.498 | |
| 最大特征向蛍中心性 Maximal eigenvector centrality | 0.29 | 0.277 | 0.238 | 0.272 | |
| 具有最大特征向量中心性的节点 Nodes with max eigenvector centrality |
OTU2614 (Hydrogenophaga) |
OTU919 (未知) |
OTU1249 (未知) |
OTU58 (Halapricum) |
|
| 特征向量中心度 Centralization of eigenvector centrality |
0.276 | 0.241 | 0.212 | 0.238 | |
| 网络密度 Graph density |
0.018 | 0.038 | 0.043 | 0.052 | |
| 效率 Efficiency |
0.943 | 0.825 | 0.882 | 0.874 | |
| 模块数 Module |
27 | 21 | 15 | 12 | |
| 模块性 Modularily |
0.736 | 0.673 | 0.737 | 0.71 | |
青藏高原的湖泊远离人类居住区, 受人类活动干扰较少, 高寒湖泊中的微生物对区域气候和环境变化敏感, 对气候和环境变化有着强烈的反馈作用[3]。盐湖是青藏高原地区主要的湖泊类型, 高原湖泊中的微生物对盐度变化有着敏感指示作用。研究青海湖和茶卡湖表层沉积物中原核微生物的分布格局与多样性特征对探索高寒湖泊生态系统的生态功能特征具有重要意义。先前研究表明盐湖的盐度[5]、pH[25]、湖底泥营养含量[26]对湖泊沉积物微生物群落起着重要的驱动作用。本研究中茶卡湖微生物群落的结构和物种多样性主要受到电导率、有效钾和碳氮比环境因子的影响, 而在青海湖中影响原核微生物群落的环境因子更多。在不同样本中, 影响原核微生物群落结构分布的因素各有差异, 这可能与优势类群的代谢方式和群体适应生存机制有关[27]。为了探讨生物和非生物因素在沉积物中微生物群落多样性的作用, 本研究用PLS-SEM分析量化了电导率、pH和沉积物的理化性质(总氮、总磷、有效氮、有效钾)对原核微生物群落多样性影响的直接和间接贡献。电导率对青海湖和茶卡湖中表层沉积物养分状态都有显著的正相关, 且在茶卡湖中, 影响程度(路径系数)增加。有效钾含量与土壤盐分高度正相关, 这与之前研究结果一致[28]。而盐湖环境较为恶劣, 其养分含量较差, 盐分的增加对总氮、有效氮、总磷含量没有显著的降低, 因此有效钾在表层沉积物营养含量中为最主要的影响因子。由于茶卡湖的理化特征变化很大, 嗜盐细菌和古菌的分布和多样性也有了较大差异。通过路径分析进一步发现环境影响因子在不同湖泊中影响原核微生物群落结构和多样性的显著性路径与正负相关关系不稳定, 若沿盐度梯度扩大采样地点、增加样本量并补充其他环境影响因子如温度、微量元素等再进一步研究, 能更好地理解原核微生物群适应极端环境的途径, 挖掘其背后的复杂机理, 揭示微生物群落组成随盐度变化的全球分布格局。本研究中茶卡湖的pH对表层沉积物营养含量有极强的正相关作用, 但青海湖和茶卡湖的pH与细菌α-多样性之间没有显著影响, 这结果也与之前研究pH在塑造湖底泥群落多样性中作用的结果一致[25], 这可能是因为影响细菌群落的结构和多样性的途径由综合变量构成, pH通过直接或间接地改变了沉积物的理化性质, 进而改变了细菌群落结构和多样性[25]。此外pH还能影响细菌的相关酶活, 影响着细菌的代谢、生长和繁殖过程[29]。茶卡湖中显著影响原核微生物群落多样性的途径较少, 可能在高盐环境下受能量限制, 微生物群落代谢和信息传递活动较少, 因此影响原核微生物群落多样性的一些随机性因素并不明显。
3.2 青海湖和茶卡湖表层沉积物中原核微生物的丰度和多样性盐湖作为高盐浓度的特殊环境, 蕴含着丰富的耐盐及嗜盐微生物资源[30]。盐度可以引起微生物对渗透调节和能量的生理约束[31], 导致微生物活性和代谢功能的潜在下降[32], 极端盐度可能对微生物生命的生存构成重大挑战。本研究中茶卡湖样本中尤其是CSe2与CSe3的群落丰度与多样性显著低于其他样本, 符合极端环境下群落多样性低的一般生态学原理[2]。QSe1—QSe3的盐度高于QSe4和QSe5, 除个别样本外, QSe1—QSe3的古菌群落丰度与多样性总体高于QSe4和QSe5。古菌群落的丰度和多样性并没有随着盐度的增加而下降, 这一发现与先前研究的古菌多样性随盐度增加而增加的结果一致[33], 这可能是因为古菌对盐胁迫具有专门的适应性, 使古菌能够在高盐浓度的环境中维持细胞功能并随着群落演替出现更多的嗜盐菌[34]。而细菌的丰度与多样性在QSe1—QSe3和QSe4—QSe5间无显著性差异, 这可归因于营养物质和资源的增加, 或由于耐盐细菌和淡水细菌在中等盐度水平下的生态位可用性增加[35]。古菌群落的丰度(Chao < 1000)与多样性(Shannon<5)显著低于细菌, 这与古菌对环境变化更为敏感, 在极端环境下的竞争优势与所受到的能量胁迫或盐度胁迫的程度以及处理这些压力时的生理适应有关[36]。此外, 样本CSe2与CSe3原核微生物过低的丰度与多样性可能是由于环境不稳定所致。CSe2的沉积物采样点在采盐航道, 易受到柴油的污染, 而CSe3的采样点在码头附近, 环境易受到机械作业等人为干扰, 原生和外来原核湖泊物种对环境变化的进化适应可能需要较长的时间。
在本研究中, Proteobacteria(34.10%)和Bacteroidetes(25.10%)是青海湖和茶卡湖共有的优势菌门, 这与He等[7]对12个青藏高原不同盐湖共有的优势菌门的结果一致, 在不同盐度(低、中、高)湖泊中, Proteobacteria和Bacteroidetes都能广泛生长。Proteobacteria和Bacteroidetes在沉积物元素循环中起着重要作用, 包括C、N、S和Fe[28]。Proteobacteria的种类和遗传多样性极其丰富, 这决定了该类群涵盖了非常广泛的代谢类型。Bacteroidetes的相对丰度沿盐度梯度相当稳定[36], 且与湖泊沉积物中的营养转化有关[37]。这两种菌门在青海湖和茶卡湖的丰富度表明它们在高寒湖泊生态系统中的生态重要性。Acetothermia为茶卡湖特有的优势菌门。以往的研究发现Acetothermia是盐湖特有菌种, 曾在死海底泥中也发现过此菌门[38], 这表明该菌门对高盐环境具有极强的适应性。本研究中古菌的Euryarchaeota(64.75%)、Woesearchaeota__DHVEG-6(27.25%)是青海湖和茶卡湖表层沉积物中原核微生物的重要组成部分。有研究发现, 盐湖以Euryarchaea为主, Euryarchaea是极端环境中常见的嗜盐古菌类群[4, 39]。相较于青海湖, Euryarchaea群落分布在茶卡湖中的表层沉积物更多。Woesearchaeota主要分布在青海湖中, 研究发现Woesearchaeota常在土壤、沉积物和其他缺氧生境等缺氧环境中占主导地位, 可能在厌氧碳循环、硫循环中发挥作用[40], 其生态学意义揭示了Woesearchaeota在特定生物群落及其在厌氧生物地球化学循环中的潜在作用。此外unclassified_k__norank主要集中在茶卡湖中, 可见茶卡湖仍存在大量未知微生物, 为了获得更多在高寒高盐环境中的原核微生物群落信息, 还需要加大采样的数量、改进宏基因组的提取方法, 加大测序广度和深度[41]。
3.3 青海湖和茶卡湖表层沉积物中原核微生物的β-多样性盐度选择微生物群落并影响微生物分布, 导致不同盐度湖泊中微生物群落组成不同[5]。在微生物的β-多样性研究中发现, 由于微生物具有较强的扩散作用, 容易形成较低的β-多样性, 但同时由于微生物具有较高的环境选择性, 也可能因此形成较高的β-多样性[42], 微生物的β-多样性变化受具体环境因素的影响。然而物种周转组分和嵌套组分及其驱动因素的相应变化仍然知之甚少[43], 特别是在高原湖泊不同盐度下微生物群落分布中。本研究发现青海湖和茶卡湖的原核微生物的总β-多样性相对较低, 这可能是因为青海湖和茶卡湖位于海拔高、气候寒冷的地区, 湖底泥养分匮乏, 生态系统脆弱。在环境选择下, 微生物群落仅限于能够在恶劣环境中生存的特定种群, 总β-多样性较低。此外总β-多样性的变化趋势与嵌套组分的变化趋势大致相同, 且物种嵌套组分对总β-多样性的贡献较大。这可能是恶劣的环境造成了扩散限制, 阻碍微生物在不同地点之间的迁移, 从而增加物种嵌套组分[8]。由于嵌套组分对空间尺度的响应不大, 当微生物群落在本地物种丰富度或/和本地个体数量上表现出高度异质性时, 嵌套组分也会起到更加重要的作用[44]。这表明青海湖和茶卡湖中原核微生物群落的适应范围小, 扩散能力弱, 遗传物质相对稳定。茶卡湖样本中CSe2和CSe3的总β-多样性和不同组分的β-多样性都低于CSe1, 这可能是人为干扰所致, 当原有生态环境遭到破坏时, 微生态环境形成不稳定状态, 而微生物具有高环境选择性, 容易形成较低的β-多样性。此外细菌和古菌的总β-多样性趋势相似, 可能是因为它们在环境选择下有着类似的生态过程[45]。在相同生境下, 原核微生物的分布格局受到一些决定性因素的影响如盐度胁迫, 从而向着一定规律的变化发展。
3.4 盐度影响湖表层沉积物中原核微生物的共生网络分子生态网络拓扑结构的变化表明, 相比青海湖, 茶卡湖原核微生物的相互作用更加精简且关系加强。这表现在:1)细菌和古菌网络中每个节点平均连接数与平均路径距离的增加;2)网络更加聚集与紧密联系;3)网络模块数量的大幅减少。因此, 茶卡湖的简单网络结构(物种之间相互作用最少)可能归因于盐饱和环境下淘汰了一些不适应环境的微生物群落, 这减少了微生物之间的直接相互作用。此外相较于茶卡湖, 青海湖的原核微生物网络结构更为复杂, 可能有以下几个原因:1)营养条件的改善有助于微生物生物量的增加, 为物种之间的相互作用提供了更多的机会;2)占据相似生态位的微生物较多, 微生物群落分布较为分散, 微生物相互作用途径更为多样化。研究发现与相对简单的网络相比, 复杂的网络具有更高的资源和信息传递效率[46], 且更高的网络复杂性导致了更稳定的微生物群落, 从而提高了它们对环境干扰的耐受性[47]。微生物群落稳定性不断提高, 对促进盐湖沉积物的物质循环具有重要意义。不同类群在青海湖和茶卡湖的分子网络表现不同。与古菌网络相比, 细菌网络的连接数和节点数更高, 细菌网络更加稳定, 面对环境因子的变化有更强的缓冲能力[48], 可见细菌和古菌在盐胁迫下的反应差异显著。
群落之间的正相关与负相关是分子网络中非常重要的特征, 并且依赖于资源可用性[49]。积极的相互作用可能反映物种之间的合作和生态位重叠, 消极的相互作用可能反映物种之间的竞争和生态位分离[50—51]。在本文的研究中, 相较于青海湖的细菌网络(54.8%)与古菌网络(64%), 茶卡湖的细菌网络(96.5%)与古菌网络(77%)物种之间积极的相互作用逐渐增加, 这表明了原核微生物在高盐环境中生态位异化较少, 在极端环境下大多数微生物物种通过相互合作以抵御恶劣的环境条件。随着生存环境和营养状况的改善, 细菌和古菌群落将扩大, 并进一步增加它们对营养的竞争。这种现象也与共存理论是一致的, 共存理论认为, 在良好的栖息地中, 高营养通量支持竞争优势的表达, 并导致劣势物种被排除在外[52]。而竞争可以促进微生物群落的网络稳定性[53], 这对增强高原盐湖生态系统的稳定性具有积极作用。这些发现也强调了在评估盐湖微生物作用的同时需考虑不同栖息地的必要性。
4 结论本研究发现表层沉积物的理化因素是影响青海湖和茶卡湖原核微生物群落组成的重要因素, 电导率是影响青海湖和茶卡湖原核微生物多样性和分布格局的最主要因素。受到盐度与环境的限制, 原核微生物扩散速度慢, 嵌套组分是原核微生物群落总β-多样性的主要驱动力, 并形成较低的总β-多样性。细菌和古菌在不同盐度环境下的响应不同, 在茶卡湖中细菌和古菌受盐度限制较大, 丰富度与多样性水平显著下降。原核微生物在青海湖和茶卡湖的群落组成差异较大。Proteobacteria和Bacteroidetes是主要优势细菌, Euryarchaeota和Woesearchaeota__DHVEG-6是主要优势古菌, 且Woesearchaeota__DHVEG-6在茶卡湖中显著减少。原核微生物在茶卡湖中存在大量耐盐和嗜盐微生物, 同时也具有更多的未知物种。本研究发现原核微生物在应对不断恶化的环境时群落之间进行更多的积极联系, 表明原核微生物具有一种通过增强合作潜力来适应环境压力的机制。因此, 在微生物保护策略中也应该考虑微生物之间的相互作用关系, 重视它们在维持生物多样性和调节生态系统服务方面的重要性, 这提高了对高寒盐湖生态系统中原核微生物的生物多样性保护策略的理解。今后应进一步完善采样规模、在更大的时空尺度上对原核微生物群落特征进行综合分析, 这有助于预测青藏高原地区湖泊群在未来环境变化下原核微生物的反应。
| [1] |
韩磊, 胡盎, 任明磊, 孟凡凡, 吴庆龙, 王建军. 湖泊微生物群落及其介导的碳循环过程. 生命科学, 2023, 35(12): 1613-1629. |
| [2] |
Shu W S, Huang L N. Microbial diversity in extreme environments. Nature Reviews Microbiology, 2022, 20(4): 219-235. DOI:10.1038/s41579-021-00648-y |
| [3] |
褚海燕. 高寒生态系统微生物群落研究进展. 微生物学通报, 2013, 40(1): 123-136. |
| [4] |
张欣, 刘静, 沈国平, 龙启福, 韩睿, 朱德锐. 基于高通量测序研究青藏高原茶卡盐湖微生物多样性. 微生物学通报, 2017, 44(8): 1834-1846. |
| [5] |
Yang J, Ma L, Jiang H C, Wu G, Dong H L. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Scientific Reports, 2016, 6: 25078. DOI:10.1038/srep25078 |
| [6] |
Ji M K, Kong W D, Yue L Y, Wang J B, Deng Y, Zhu L P. Salinity reduces bacterial diversity, but increases network complexity in Tibetan Plateau lakes. FEMS Microbiology Ecology, 2019, 95(12): fiz190. DOI:10.1093/femsec/fiz190 |
| [7] |
He Y Y, He L L, Wang Z, Liang T, Sun S C, Liu X S. Salinity shapes the microbial communities in surface sediments of salt lakes on the Tibetan Plateau, China. Water, 2022, 14(24): 4043. DOI:10.3390/w14244043 |
| [8] |
斯幸峰, 赵郁豪, 陈传武, 任鹏, 曾頔, 吴玲兵, 丁平. Beta多样性分解: 方法、应用与展望. 生物多样性, 2017, 25(5): 464-480. |
| [9] |
Baselga A. Partitioning the turnover and nestedness components of beta diversity. Global Ecology and Biogeography, 2010, 19(1): 134-143. DOI:10.1111/j.1466-8238.2009.00490.x |
| [10] |
Wu L B, Si X F, Didham R K, Ge D P, Ding P. Dispersal modality determines the relative partitioning of beta diversity in spider assemblages on subtropical land-bridge islands. Journal of Biogeography, 2017, 44(9): 2121-2131. DOI:10.1111/jbi.13007 |
| [11] |
Si X F, Baselga A, Ding P. Revealing Beta-diversity patterns of breeding bird and lizard communities on inundated land-bridge islands by separating the turnover and nestedness components. PLoS ONE, 2015, 10(5): e0127692. DOI:10.1371/journal.pone.0127692 |
| [12] |
Li X R, Liu Q, Yu X W, Zhang C R, Liu M J, Zhou X H, Gu C X, Wang M, Shao H B, Li J S, Jiang Y. Spatial pattern and co-occurrence network of microbial community in response to extreme environment of salt lakes on the Qinghai-Tibet Plateau. Environmental Science and Pollution Research, 2023, 30(8): 20615-20630. |
| [13] |
Faust K, Raes J. Microbial interactions: from networks to models. Nature Reviews Microbiology, 2012, 10(8): 538-550. DOI:10.1038/nrmicro2832 |
| [14] |
石文莉, 蒋如东, 马天海, 阮晓红, 张亚平, 白莹. 太湖不同营养水平湖区沉积环境微生物分子生态网络特征及其环境响应分析. 南京大学学报, 自然科学, 2018, 54(5): 1045-1056. |
| [15] |
张佩莲, 张含笑, 霍守亮, 李怡, 张靖天, 翁南燕. 青藏高原不同盐度湖泊细菌群落多样性及DOM分布特征. 环境科学学报, 2024, 44(5): 158-168. |
| [16] |
Yang G Q, Zhang M, Xie Z H, Li J Y, Ma M G, Lai P Y, Wang J B. Quantifying the contributions of climate change and human activities to water volume in Lake Qinghai, China. Remote Sensing, 2022, 14(1): 99. |
| [17] |
Huang J R, Yang J, Jiang H C, Wu G, Liu W, Wang B C, Xiao H Y, Han J B. Microbial Responses to Simulated Salinization and Desalinization in the Sediments of the Qinghai-Tibetan Lakes. Frontiers in Microbiology, 2020, 11: 1772. DOI:10.3389/fmicb.2020.01772 |
| [18] |
刘冉, 赵海波, 王冰玥, 栗冠媛, 吴红. 浅谈电导率仪的发展和计量方法的研究. 分析仪器, 2021(3): 112-116. |
| [19] |
范成新. 湖泊沉积物调查规范. 北京: 科学出版社, 2018.
|
| [20] |
鲍士旦. 土壤农化分析. 北京: 中国农业出版社, 2000.
|
| [21] |
Henseler J, Hubona G, Ray P A. Using PLS path modeling in new technology research: updated guidelines. Industrial Management & Data Systems, 2016, 116(1): 2-20. |
| [22] |
Deng Y, Jiang Y H, Yang Y F, He Z L, Luo F, Zhou J Z. Molecular ecological network analyses. BMC Bioinformatics, 2012, 13: 113. DOI:10.1186/1471-2105-13-113 |
| [23] |
Shannon P, Markiel A, Ozier O, Baliga N S, Wang J T, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 2003, 13(11): 2498-2504. DOI:10.1101/gr.1239303 |
| [24] |
Baselga A, Orme C D L. Betapart: an R package for the study of beta diversity. Methods in Ecology and Evolution, 2012, 3(5): 808-812. DOI:10.1111/j.2041-210X.2012.00224.x |
| [25] |
Pu H G, Yuan Y X, Qin L, Liu X H. pH drives differences in bacterial community β-diversity in hydrologically connected lake sediments. Microorganisms, 2023, 11(3): 676. |
| [26] |
Han X G, Schubert C J, Fiskal A, Dubois N, Lever M A. Eutrophication as a driver of microbial community structure in lake sediments. Environmental Microbiology, 2020, 22(8): 3446-3462. |
| [27] |
朱德锐, 韩睿, 石晴, 沈国平, 龙启福, 双杰. 青藏高原盐湖细菌群落与超盐环境因素的相关性. 中国环境科学, 2017, 37(12): 4657-4666. |
| [28] |
Wang N N, Qi W, Wang D, Qin T T, Lu C. Spatial variability of soil nutrients and salinity in coastal saline-alkali land based on belt transect method. Ying Yong Sheng Tai Xue Bao, 2012, 23(6): 1527-1532. |
| [29] |
Rousk J, Baath E, Brookes P C, Lauber C L, Lozupone C, Caporaso J G, Knight R, Fierer N. Soil bacterial and fungal communities across a pH gradient in an arable soil. The ISME Journal, 2010, 4(10): 1340-1351. |
| [30] |
李二阳, 马雪莉, 吕杰, 马媛, 吕光辉. 新疆天山北坡不同盐湖微生物菌群结构及其影响因子. 生态学报, 2021, 41(18): 7212-7225. DOI:10.5846/stxb202010122601 |
| [31] |
Yang J, Jiang H C, Liu W, Huang L Q, Huang J R, Wang B C, Dong H L, Chu R K, Tolic N. Potential utilization of terrestrially derived dissolved organic matter by aquatic microbial communities in saline lakes. The ISME Journal, 2020, 14(9): 2313-2324. |
| [32] |
Pavloudi C, Kristoffersen J B, Oulas A, De Troch M, Arvanitidis C. Sediment microbial taxonomic and functional diversity in a natural salinity gradient challenge Remane's "species minimum" concept. PeerJ, 2017, 5: e3687. |
| [33] |
Liu Y Q, Priscu J C, Xiong J B, Conrad R, Vick-Majors T, Chu H Y, Hou J Z. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiology Ecology, 2016, 92(3): fiw033. |
| [34] |
Valentine D L. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nature Reviews Microbiology, 2007, 5(4): 316-323. |
| [35] |
Wang J J, Yang D M, Zhang Y, Shen J, van der Gast C, Hahn M W, Wu Q L. Do patterns of bacterial diversity along salinity gradients differ from those observed for macroorganisms?. PLoS ONE, 2011, 6(11): e27597. |
| [36] |
Zhong Z P, Liu Y, Miao L L, Wang F, Chu L M, Wang J L, Liu Z P. Prokaryotic community structure driven by salinity and ionic concentrations in plateau lakes of the Tibetan Plateau. Applied and Environmental Microbiology, 2016, 82(6): 1846-1858. |
| [37] |
Song H, Li Z, Du B, Wang G, Ding Y. Bacterial communities in sediments of the shallow Lake Dongping in China. Journal of Applied Microbiology, 2012, 112(1): 79-89. |
| [38] |
Thomas C, Ariztegui D. Fluid inclusions from the deep Dead Sea sediment provide new insights on Holocene extreme microbial life. Quaternary Science Reviews, 2019, 212: 18-27. |
| [39] |
Mwirichia R, Cousin S, Muigai A W, Boga H I, Stackebrandt E. Archaeal diversity in the haloalkaline Lake Elmenteita in Kenya. Current Microbiology, 2010, 60(1): 47-52. |
| [40] |
Liu X B, Wang Y L, Gu J D. Ecological distribution and potential roles of Woesearchaeota in anaerobic biogeochemical cycling unveiled by genomic analysis. Computational and Structural Biotechnology Journal, 2021, 19: 794-800. |
| [41] |
薛凯, 张彪, 周姝彤, 冉沁蔚, 唐立, 车荣晓, 庞哲, 王芳, 王頔, 张静, 姜丽丽, 胡容海, 崔骁勇, 郝彦宾, 王艳芬. 青藏高原高寒草地土壤微生物群落及影响因子. 科学通报, 2019, 64(27): 2915-2927. |
| [42] |
Wang J J, Pan F Y, Soininen J, Heino J, Shen J. Nutrient enrichment modifies temperature-biodiversity relationships in large-scale field experiments. Nature Communications, 2016, 7: 13960. |
| [43] |
Wu K Y, Zhao W Q, Li M J, Picazo F, Soininen J, Shen J, Zhu L F, Cheng X Y, Wang J J. Taxonomic dependency of beta diversity components in benthic communities of bacteria, diatoms and chironomids along a water-depth gradient. Science of The Total Environment, 2020, 741: 140462. |
| [44] |
Wang X G, Wiegand T, Anderson-Teixeira K J, Bourg N A, Hao Z Q, Howe R, Jin G Z, Orwig D A, Spasojevic M J, Wang S Z, Wolf A, Myers J A. Ecological drivers of spatial community dissimilarity, species replacement and species nestedness across temperate forests. Global Ecology and Biogeography, 2018, 27(5): 581-592. |
| [45] |
Shade A, Dunn R R, Blowes S A, Keil P, Bohannan B J M, Herrmann M, Kusel K, Lennon J T, Sanders N J, Storch D, Chase J. Macroecology to unite all life, large and small. Trends in Ecology & Evolution, 2018, 33(10): 731-744. |
| [46] |
Wagg C, Schlaeppi K, Banerjee S, Kuramae E E, van der Heijden M G A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nature Communications, 2019, 10: 4841. |
| [47] |
de Vries F T, Griffiths R I, Bailey M, Craig H, Girlanda M, Gweon H S, Hallin S, Kaisermann A, Keith A M, Kretzschmar M, Lemanceau P, Lumini E, Mason K E, Oliver A, Ostle N, Prosser J I, Thion C, Thomson B, Bardgett R D. Soil bacterial networks are less stable under drought than fungal networks. Nature Communications, 2018, 9: 3033. |
| [48] |
汪峰, Zhou J Z, 孙波. 我国东部土壤氮转化微生物的功能分子生态网络结构及其对作物的响应. 科学通报, 2014, 59(Z1): 387-396. |
| [49] |
Zhao D Y, Shen F, Zeng J, Huang R, Yu Z B, Wu Q L. Network analysis reveals seasonal variation of co-occurrence correlations between Cyanobacteria and other bacterioplankton. Science of The Total Environment, 2016, 573: 817-825. |
| [50] |
Deng Y, Zhang P, Qin Y J, Tu Q C, Yang Y F, He Z L, Schadt C W, Zhou J Z. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environmental Microbiology, 2016, 18(1): 205-218. |
| [51] |
Ghoul M, Mitri S. The ecology and evolution of microbial competition. Trends in Microbiology, 2016, 24(10): 833-845. |
| [52] |
储诚进, 王酉石, 刘宇, 蒋林, 何芳良. 物种共存理论研究进展. 生物多样性, 2017, 25(4): 345-354. |
| [53] |
Coyte K Z, Schluter J, Foster K R. The ecology of the microbiome: networks, competition, and stability. Science, 2015, 350(6261): 663-666. |