 2024, Vol. 44
2024, Vol. 44文章信息
- 马馨怡, 高威, 李颜, 郭晓彬, 贾仲君, 吴金水, 王连峰
- MA Xinyi, GAO Wei, LI Yan, GUO Xiaobin, JIA Zhongjun, WU Jinshui, WANG Lianfeng
- 基于细胞水平高通量测序的土壤水分对活性微生物的影响研究
- Effect of soil water status on active microbes at cellular level based on high-throughput sequencing
- 生态学报. 2024, 44(16): 7160-7171
- Acta Ecologica Sinica. 2024, 44(16): 7160-7171
- http://dx.doi.org/10.20103/j.stxb.202309252082
-
文章历史
- 收稿日期: 2023-09-25
- 网络出版日期: 2024-06-18






2. 中国科学院亚热带农业生态研究所亚热带农业生态过程重点实验室, 长沙 410125;
3. 中国科学院南京土壤研究所土壤与农业可持续发展国家重点实验室, 南京 210008
2. Key Laboratory of Agro-ecological Processes in Subtropical Region, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha 410125, China;
3. State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China
《东北黑土地白皮书(2020)》显示, 东北黑土地总面积109万km2, 是我国最重要的商品粮基地, 被誉为国家粮食安全的“稳压器”和“压舱石”。黑土以高有机质著称, 是地球上最肥沃的土壤之一。然而, 因长期过度开发利用、气候变化等多种因素的影响, 东北黑土地出现了不同程度退化问题, 直接影响到区域粮食产量的稳定[1]。因此, 如何提升黑土功能稳定性是最重要的科学问题[1—2]。土壤水分是影响黑土功能稳定性的重要因素之一。土壤水分变化通过改变土壤养分的物理扩散和化学形态转化等过程, 影响了土壤微生物区系和功能活性, 最终影响土壤养分循环过程[3—4]。在全球气候变暖的背景下, 位于半湿润气候带的东北黑土区易发生极端降雨和干旱, 导致土壤水分含量的频繁波动。因此, 研究不同水分状况下黑土活性微生物群落结构变化对黑土区合理水分管理具有重要意义。
认识土壤水分对土壤微生物活性的影响是深入解析不同水分状况下土壤养分迁移转化的重要前提。土壤水分含量的变化一方面通过改变土壤可溶性养分基质的运移, 影响土壤微生物呼吸活性和微生物群落结构, 进而影响土壤元素循环过程[5], 另一方面土壤水分变化直接改变了土壤氧化还原状况, 改变了土壤微生物群落结构特征, 进而对土壤养分累积和碳氮功能转化造成影响[3—4]。早在1959年有研究发现, 经过干旱的土壤在重新湿润后, 土壤有机碳矿化速率在数小时内迅速增加, 造成土壤有机碳的大量流失[6]。也有研究发现, 高土壤水分状况下由于改变了土壤碳代谢的微生物过程[7], 土壤微生物呼吸明显减弱。然而, 由于土壤空间变异大和微生物类群多样, 使得土壤碳氮功能变化和微生物呼吸过程对水分条件变化的响应研究变得更加复杂, 其响应机制仍需进一步探索。
土壤微生物被称为土壤碳氮养分循环的引擎, 其活性和群落结构很大程度上决定了土壤肥力状况[8—9]。近年来, 随着高通量测序技术和生物信息学的飞速发展, 土壤微生物研究已经成为土壤学研究的常规手段之一[10]。土壤水分是影响土壤微生物群落结构变化的重要因子之一[11—12]。Li等人的研究中发现, 降水改变土壤水分有效性进而影响土壤微生物群落结构, 且降水减少对土壤微生物数量存在明显的抑制作用[13—14]。由于土壤水分在时空分布上的异质性和不确定性, 使得土壤微生物群落与水分条件变化关系的研究变得困难, 确实已有研究通过土壤水分梯度控制培养试验来研究土壤水分变化对土壤微生物群落的影响[15—16]。但是, 当前的研究更多地基于土壤总微生物类群而非重要的活性功能微生物类群。考虑到土壤中仅有少量的微生物具有活性[17], 如果仅从土壤总微生物群落水平解析土壤水分对土壤微生物功能过程的影响研究会引起一定的偏差。此外, 传统的平板计数法以及现代的DNA-SIP技术方法均可以在一定程度上鉴定土壤活性微生物[18—19], 但是平板计数主要针对少量的可培养微生物[18], 而DNA-SIP成本高, 操作复杂, 技术难度大[20]。据此, 本研究通过经典的土壤细胞提取方法, 并结合先进的高通量测序技术, 理论上可以较好的定量研究土壤活性微生物。但是, 在细胞水平研究复杂土壤活性微生物群落及其功能过程尚不多见。有研究结果表明[19], 基于细胞水平的高通量测序能够较好地表征水稻土甲烷氧化的微生物功能转化过程。
据此, 本实验采用微宇宙控制试验, 以东北典型水稻土为研究对象, 动态监测土壤微生物呼吸活性, 结合土壤微生物细胞提取技术以及高通量测序技术, 在土壤总微生物(土壤DNA)和活性微生物(细胞DNA)水平解析了两种水分(湿润和淹水)状况下土壤微生物群落结构的响应特征。研究结果为深入理解水分变化下土壤微生物活性及其功能演变过程提供依据, 对黑土区稻田合理水分管理具有重要借鉴意义。
1 材料与方法 1.1 土壤样品概况供试土壤样品采集自辽宁省锦州市水田(121°30′ E, 41°30′ N)。研究区为温带半湿润大陆性季风气候, 年均气温9.0 ℃, 年最高温41.8 ℃, 年最低温-31.3 ℃, 年降水量450—580 mm。供试土壤类型为东北水稻土。通过蛇形法采集0—20 cm表层新鲜土壤, 混合均匀, 除去肉眼可见的植物细根及杂物, 研磨过2 mm筛。在进行微宇宙培养试验前测定了土壤基本理化性质, 所用测定方法参见文献[21]。具体地, 取10 g新鲜土壤放置于105 ℃烘箱中烘至恒重后测定土壤含水量为(13.5±0.43)%;采用流动分析仪测定土壤无机氮含量, 其中土壤铵态氮(NH4+-N)(为5.03±0.22) mg/kg, 土壤硝态氮(NO3--N)为(3.31±0.39) mg/kg。另取部分样品风干, 测定土壤pH(H2O)为7.18±0.07(水土比为2.5 ∶ 1);采用碳氮元素分析仪(Vario Max CN, Germany)测定土壤碳氮全量, 其中全碳(TC)为(8.73±0.25) g/kg, 土壤全氮(TN)为(0.91±0.05) g/kg。
1.2 土壤微宇宙培养微宇宙培养试验操作流程。首先称取相当于10 g干土的新鲜土样置于120 mL血清瓶, 调节土壤水分含量, 设置三个处理, 包括:(1)对照处理(调节土壤含水量为60% WHC, 不加任何碳氮底物);(2)湿润处理(调节土壤含水量为60% WHC, 并加入5 mg C-乙酸钠和1 mg N-硝酸钾的碳氮底物);(3)淹水处理(调节土壤含水量为100% WHC, 并加入5 mg C-乙酸钠和1 mg N-硝酸钾的碳氮底物)。水分调节完成后, 通过高纯N2反复冲刷培养瓶3次后迅速用丁基胶塞密封, 然后通过抽真空气体置换的方法, 向培养瓶注入同等体积的高纯空气, 以保持培养瓶内外气压平衡。最后, 放恒温25 ℃培养箱避光培养48 h。培养过程中, 动态监测培养瓶顶空CO2排放量, 随后立即置换高纯N2。培养结束后提取土壤总DNA及土壤细胞DNA, 进行后续的高通量测序与分析。
1.3 土壤微生物呼吸活性的测定土壤CO2排放通量是表征土壤微生物呼吸活性的重要指标。在培养0 h, 6 h, 12 h, 18 h, 24 h, 36 h, 48 h动态监测培养瓶气体中CO2浓度。CO2通过气相色谱仪(Agilent 7890, USA)分析测定。CO2累积排放量的计算公式为:
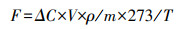
|
(1) |
式中, F为CO2累积排放量(μg/g), ΔC为CO2浓度的变化量(μg C/g);V为培养瓶体积(L);ρ为标准状态下CO2密度(g/L);m为干土质量(g);T为培养温度(K)。
1.4 土壤总DNA和细胞DNA提取土壤总DNA提取。本实验主要采用Fast DNA Spin Kit for Soil(MP Biomedicals公司)试剂盒提取土壤总DNA, 具体参考试剂盒操作指南。土壤总DNA的质量和纯度通过微量紫外分光光度计(NanoDrop ND-1000, USA)测定。提取的土壤DNA, 保存至-80 ℃, 用于后续高通量测序分析。
土壤细胞DNA提取。利用差速离心结合Nycodenz密度梯度离心的方法从土壤中提取活性微生物细胞[19], 具体步骤如下:首先, 称取1 g土壤样本至无菌离心管中, 加入玻璃珠和0.2%焦磷酸钠溶液5 mL后离心。然后, 将上清液与4 mL Nycodenz溶液充分混合再次离心。转移灰白细胞层, 用缓冲液洗涤细胞, 0.5 mL多聚甲醛重悬细胞沉淀, 4 ℃固定保存。提取土壤活性微生物细胞后, 将细胞悬液过滤到孔径0.22 μm的聚碳酸酯滤膜上, 用无菌剪刀将滤膜剪碎, 收集至无菌离心管中, 提取细胞DNA用于高通量测序分析。
1.5 实时荧光定量PCR、Miseq高通量测序和生物信息学分析DNA提取后通过16S rRNA基因的实时荧光定量PCR(qPCR)表征土壤微生物数量。16S rRNA基因定量的通用引物为515F(GTGCCAGCMGCCGCGGTAA)和907R(CCGTCAATTCMTTTRAGTTT)[22], 具体操作参见文献[19]。16S rRNA基因qPCR的扩增效率为98.5%。进一步通过16S rRNA基因的Miseq高通量测序分析土壤微生物群落结构。16S rRNA基因V4—V5区扩增通用引物为515F/907R。扩增反应体系为:Taq PCR Master Mix 9 μL, 模板DNA 1 μL, 上下游引物0.25 μL以及无菌蒸馏水16.5 μL。扩增程序为:(1)94 ℃预变性5 min;(2)94 ℃变性30 s, 55 ℃退火45 s, 72 ℃延伸1 min, 持续32个循环;(3)72 ℃最终延伸10 min, 降温至4 ℃后实施扩增。扩增结束后用2%琼脂糖凝胶电泳检查PCR产物质量, 切胶回收目的片段, 经过纯化后等质量混合, 进行建库和测序分析。
高通量测序数据经过序列拼接、过滤并去除嵌合体和短序列等质控过程后共计得到562, 929条16S rRNA高质量序列。高通量测序数据分析主要借助QIIME软件完成[23—24]。简要来说, 首先通过“join_paired_ends.py”指令合并双端测序文件, 通过“extract_barcodes.py”指令去除测序接头, 然后去除序列片段长度 < 200 bp, 质量控制参考值< 25, 以及无法匹配的低质量序列, 并采用Usearch软件去除嵌合体, 并以97%的相似度将序列聚类为操作分类单元(OTUs), 最后与RDP数据库(http://rdp.cme.msu.edu)进行比对, 最终得到每个样品的物种组成。进一步, 基于16S的OTU分类注释表比对至FAPROTAX(functional annotation of prokaryotic taxa)功能注释数据库得到不同水分状况下土壤微生物生态功能丰度[25]。
微生物多样性主要通过Alpha和Beta多样性指数构建。多样性分析之前要进行稀释曲线的绘制来表征测序深度的合理性, 间接反映样品中物种丰富程度。常用的Alpha指数, 如Chao1指数(丰富度)和Shannon指数(均匀度)等, 是反映物种丰富度和均匀度的综合指标, 具体通过“collate alpha”指令完成。而Beta多样性主要表征不同环境群落之间的物种差异性, 主要通过PCoA(Principal Coordinates Analysis)反应微生物群落组成的差异情况, PCoA的计算主要通过R语言Vegan软件包完成。
1.6 微生物共存生态网络构建在微生物网络构建前, 先剔除微生物类群(OTUs)丰度低于0.06%的低丰度类群。利用R语言的corr.test函数计算OTUs之间的Spearman相关系数, 得到相关性系数矩阵和P值矩阵, Spearman相关系数和P值的阈值分别设定为0.7和0.05。网络分析将系统中的每一个菌群看作一个节点(Node), 利用节点间两两相关形成的边构建起网络结构, 以体现整体系统及个别拓扑性质, 最后通过Gephi软件进行可视化分析[26]。
1.7 统计分析所有数据处理均通过Microsoft Excel 2019、R 4.2.0和SPSS 26.0完成。采用SPSS 26.0软件包(SPSS Inc., Cary, NC, USA)中的one-way ANOVA进行统计显著性检验, P < 0.05表示差异显著。微生物共存网络可视化由Gephi 0.9.2完成, 其他图表均通过Origin软件(OriginPro 2021, 美国)进行绘制。
2 结果与分析 2.1 不同水分状况对东北水稻土微生物呼吸活性和多样性的影响土壤微生物呼吸是表征土壤微生物活性的重要指标。结果表明, 水分增加显著抑制了土壤微生物呼吸活性(图 1)。在培养48 h, 土壤CO2累积排放量由对照处理的80.8 μg C/g显著提升到了湿润处理的175.8 μg C/g和淹水处理的153.1 μg C/g, 分别增加了1.18倍和0.89倍。淹水环境土壤微生物呼吸活性显著低于湿润处理, 两者相差1.13倍。进一步通过土壤总DNA和细胞DNA的高通量测序表征了土壤总微生物和活性微生物类群数量和多样性。16S rRNA基因定量结果表明, 土壤水分增加显著增加了土壤总微生物和活性微生物数量(图 1)。土壤总微生物细菌数量由湿润处理的1.26×1011个/g干土增加至淹水处理的1.54×1011个/g干土, 增幅为22.2%。而土壤活性微生物细菌数量由湿润处理的1.78×109个/g干土增加至淹水处理的2.35×109个/g干土, 增幅为32.1%。此外, 土壤总微生物细菌数量远高于土壤活性微生物细菌数量, 两者相差最高可达70倍, 表明土壤中仅有少量的微生物具有活性。然而, 土壤水分对土壤总微生物和活性微生物水平的多样性影响显著(图 1), 土壤总微生物Shannon指数由湿润处理的3.56增加至淹水处理的4.15, 增幅为17.0%, 而土壤活性微生物Shannon指数由湿润处理的5.36减至淹水处理的4.29, 降幅达20.0%。PCoA也进一步证实了土壤水分显著影响了土壤总微生物与活性微生物群落结构(图 1)。

|
| 图 1 不同水分条件下土壤CO2累积排放量、16S rRNA基因拷贝数、Shannon指数和主坐标分析(PCoA) Fig. 1 Cumulative soil CO2 emissions, 16S rRNA gene copies, Shannon index and PCoA analysis under different water status in paddy soil CK:对照处理;MS:湿润处理;FS:淹水处理 |
不同水分状况导致土壤微生物群落组成发生显著变化(图 2)。在微生物门水平上, 不同水分条件下土壤活性微生物类群和总微生物类群门水平较为一致(图 2)。不同水分状况下土壤总微生物和活性微生物共性的微生物门高达7个, 其占总微生物类群的丰度超过95.0%。不同微生物门对土壤水分的响应具有显著差异(图 2)。在土壤总微生物水平, 湿润处理共检测出7个门, 优势门分别为变形菌门(Proteobacteria, 62.4%)、厚壁菌门(Firmicutes, 22.7%)以及绿湾菌门(Chloroflexi, 6.1%)。而淹水处理检测出的微生物门与湿润处理一致, 但是微生物类群丰度具有差异。例如, 淹水处理变形菌门(53.4%)相比于湿润处理相对丰度降低了16.9%, 而淹水处理厚壁菌门(26.3%)和绿湾菌门(7.4%)与湿润处理相比分别增加了3.6%和1.3%。土壤活性微生物类群也发现类似的趋势。在高水分淹水环境下, 优势活性微生物变形菌门的相对丰度由46.0%后降至湿润处理的42.5%, 微生物丰度降低了3.5%。而活性微生物厚壁菌门的相对丰度由21.8%显著增加至湿润处理的34.6%, 微生物丰度增幅高达58.7%, 绿湾菌门也增加了16.3%。

|
| 图 2 不同水分条件下土壤微生物门数量和门相对丰度、微生物属数量和属相对丰度 Fig. 2 Number and abundance of soil microbes at phylum and genus levels under different water status in paddy soil Armatimonadetes:装甲菌门;Planctomycetes:浮霉菌门;Cyanobacteria/Chloroplast:蓝藻门;Acidobacteria:酸杆菌门;Bacteroidetes:拟杆菌门;Unclassified_Bacteria:未分类的细菌;Chloroflexi:绿湾菌门;Actinobacteria:放线菌门;Firmicutes:厚壁菌门;Proteobacteria:变形菌门;Unclassified_Clostridiales:未分类的梭状芽孢杆菌;Unclassified_Burkholderiales:未分类的伯克霍尔德菌;Desulfocapsa:硫化功能菌;Azotobacter:节杆菌;Arthrobacter:节杆菌;Unclassified_Anaerolineaceae:未分类的厌氧绳菌;Unclassified_Firmicutes:未分类的厚壁菌;Sporosarcina:八叠球菌;Pseudomonas:假单胞菌 |
在高精细化分类微生物属水平, 不同水分条件下活性微生物类群差异更加明显(图 2)。土壤总微生物水平, 对照、湿润和淹水处理分别检测386、372和366属。湿润、淹水状况与对照处理共有属占比分别高达93.5%和92.9%。类似的, 土壤活性微生物中湿润、淹水状况与对照处理共有属占比也分别高达92.7%和93.0%(图 2)。然而, 不同微生物属对土壤水分的响应具有显著差异(图 2)。例如, 针对土壤总微生物属, 湿润处理优势属为假单胞菌(Pseudomonas, 38.4%)、固氮菌属(Azotobacter, 8.5%)和八叠球菌(Sporosarcina, 3.4%), 而在淹水处理其相对丰度分别为31.9%、5.7%和2.2%, 其微生物丰度分别降低了16.9%、32.9%和35.3%。针对土壤活性微生物属, 优势假单胞菌随水分含量增加而显著增加, 其占比由湿润处理的6.7%显著升高至淹水处理的11.6%, 两者相差1.73倍。同一水分状况下, 具体的微生物功能类群在土壤总微生物和土壤活性微生物中的相对丰度也具有显著差异。例如, 在湿润条件下, 优势微生物假单胞菌在土壤总微生物中相对丰度为38.4%, 但其在土壤活性微生物中仅占6.7%。类似地, 在淹水条件下假单胞菌在土壤总微生物中相对丰度高达31.9%, 但在土壤活性微生物中仅占11.6%。这暗示着基于土壤总微生物群落水平的微生物研究可能会高估土壤微生物的功能活性。
2.3 不同水分状况下东北水稻土活性微生物的比较分析进一步通过两种方法来鉴别土壤活性的微生物类群。方法(1):基于土壤总微生物类群分析。与对照相比, 土壤微生物丰度发生显著性增加的微生物类群被认为具有活性。方法(2):基于土壤活性微生物分析。直接提取土壤微生物细胞DNA并进行高通量测序, 这部分类群认为具有活性, 且微生物丰度越高, 其活性也越强。
不同水分状况下优势的活性微生物类群组成相似, 但微生物丰度发生了显著变化(图 3)。基于土壤细胞的活性微生物组分析, 湿润状况下优势的活性微生物为假单胞菌(Pseudomonas, 6.7%)、固氮菌(Arthrobacter, 6.9%)、八叠球菌(Sporosarcina, 4.9%)、未分类的伯克霍尔德菌(Unclassified_Burkholderiales, 4.7%)和藤黄单胞菌(Luteimonas, 4.0%), 其仅占总活性微生物类群的27.2%。而淹水状况下优势的活性微生物类群更加富集, 主要为假单胞菌(Pseudomonas, 11.6%)、固氮菌(Arthrobacter, 7.6%)、八叠球菌(Sporosarcina, 7.6%)、未分类的伯克霍尔德菌(Unclassified_Burkholderiales, 4.2%)和藤黄单胞菌(Luteimonas, 0.05%), 其占总活性微生物类群的比例高达31.05%。而基于DNA测序的活性微生物类群分析发现, 与对照相比, 湿润状况下土壤微生物增幅最大的微生物类群主要为假单胞菌属(Pseudomonas, 38.4%)、固氮菌属(Azotobacter, 8.5%)和芽孢杆菌属(Bacillus, 6.3%), 其占总微生物类群的53.2%;而淹水状况下增幅最大的活性微生物类群主要为假单胞菌属(Pseudomonas, 31.9%)、固氮菌属(Azotobacter, 5.7%)和芽孢杆菌属(Bacillus, 1.7%), 其占总微生物类群的39.3%。基于土壤总微生物和土壤细胞活性微生物对比分析发现, 基于土壤总微生物中鉴定活性微生物的策略对土壤中优势的微生物类群较为准确, 而在数量上占弱势的微生物类群中并没有得到很好的效果。总之, 尽管这两种活性微生物分析策略的分辨率并不相同, 但均较好地表征了土壤中微生物丰度占优势的活性微生物类群。

|
| 图 3 不同水分条件下土壤主要的活性微生物类群 Fig. 3 Soil dominant active microbes under different water status in paddy soil 柱状图表示土壤活性微生物相对丰度, 点状图表示与对照相比土壤总微生物相对丰度的净增加量 |
微生物共存网络的构建是深入理解不同环境下土壤微生物群落间及其相关关系的重要手段。本研究构建了不同水分条件下土壤微生物共存网络。高水分淹水处理会导致土壤微生物关联网络更加松散, 显著减少土壤微生物之间的正向相关关系, 强化微生物群落之间的竞争作用(图 4)。具体地, 低水分湿润处理共计发现5489条共存网络关联曲线, 其中正向关联网络占比高达为86.2%(4729/5489)。相比之下, 高水分淹水处理共计发现2388条共存网络关联曲线, 正向关联网络比例显著降低, 仅为61.4%(1466/2388)。而优势的关键微生物类群假单胞菌也发现类似的结果。湿润处理共计发现61条假单胞菌与其他微生物的显著关联关系, 相比较而言, 淹水处理共存网络显著的关联数仅为15。这表明, 低水分管理模式显著增加了土壤微生物间的正向相关关系, 进而更有利于土壤微生物的共存。

|
| 图 4 不同水分条件下土壤活性微生物和关键物种Pseudomonas相关类群的微生物共存网络 Fig. 4 The network analysis showing the co-occurrence patterns of soil active microbes and Pseudomonas-centered phylotypes under different water status in paddy soil |
采用FAPROTAX对不同水分状况下东北水稻土细菌群落进行了功能预测分析。结果表明, 化能异养(主要为好氧化能异养)、生物固氮、硫化合物呼吸代谢是不同水分状况下东北水稻土主要的地球化学元素循环过程(图 5)。但是不同水分状况对土壤总微生物和活性微生物功能影响差异显著。在土壤总微生物水平, 好氧化能异养过程(主要的化能异养过程)占比由湿润处理的36.6%降至淹水处理的33.5%。同样, 土壤活性微生物中好氧化能异养过程占比由湿润处理的26.2%降至淹水处理的22.9%。相反的, 硫化合物呼吸过程和硫酸盐呼吸过程的表达量随土壤水分升高而逐渐升高。硫化合物呼吸过程在土壤总微生物水平由湿润处理的1.9%升至淹水处理的2.5%, 而土壤活性微生物水平由湿润处理的6.7%显著升高至淹水处理的13.0%。硫酸盐呼吸过程在土壤总微生物水平由湿润处理的1.6%升至淹水处理的2.2%, 而土壤活性微生物水平由湿润处理的6.5%显著升高至淹水处理的12.8%。不同水分状况通过改变了微生物群落和区系特征, 进而影响了土壤元素循环和微生物代谢过程。

|
| 图 5 不同水分条件下土壤细菌功能预测 Fig. 5 Prediction of soil bacterial function under different water conditions |
通过提取土壤DNA和土壤细胞悬液DNA进行高通量测序的方法来开展土壤水分对土壤活性微生物的影响研究。结果表明, 尽管土壤DNA和细胞DNA水平在微生物组研究方面的分辨率具有差异, 但是均能较好反应土壤中数量上占优势的活性功能微生物类群。与湿润处理相比, 淹水处理显著降低了土壤微生物呼吸活性, 改变了土壤微生物群落结构和多样性, 增加了土壤微生物数量, 强化土壤微生物群落之间的竞争作用, 进而影响了土壤微生物功能过程。
土壤微生物呼吸是表征土壤质量和土壤肥力的重要生物学指标, 反映了土壤微生物活性和物质代谢的强度[27]。土壤水分是影响土壤物质运移和土壤氧化还原状况的重要因子[28]。高水分显著抑制了土壤微生物呼吸活性(图 1), 这可能是由于水分的增加堵塞了土壤孔隙, 可利用氧气减少, 降低了土壤有机碳的矿化速率与土壤CO2的排放速率, 使高水分含量下土壤总微生物呼吸活性减弱。这与孙中林等[29]研究发现淹水条件不利于土壤有机碳矿化的结果类似。此外, 微生物类群和多样性也会显著影响土壤有机碳矿化。不同的水势条件下, 土壤中微生物类群和活性的改变会显著影响土壤微生物呼吸活性[30—31]。我们也得到类似的结果, 高水分状况显著改变了土壤活性微生物群落结构和多样性, 进而可能会影响土壤有机碳的矿化。土壤微生物群落的改变反映了土壤微生物的适应能力[30]。土壤总微生物多样性随着土壤水分含量的增加而增加(图 1), 这主要是因为在土壤水分含量变化过程中, 土壤中不同的微生物种群具有不同的生长代谢速率和适应能力[3, 7]。水稻土长期处于高水分环境, 土壤氧化还原电位较低, 抑制了土壤中好氧微生物的生长[14], 而某些厌氧或微好氧微生物长期适应淹水环境可能繁殖较快并逐渐成为土壤微生物群落的优势种群。值得注意的是, 土壤活性微生物多样性随着水分增加反而减少, 这可能由于淹水状况增加了土壤活性微生物间的负相关关系, 强化了活性微生物间的竞争关系, 进而导致活性微生物多样性的减少。稻田生态系统中活性微生物在短期内对高水分环境的强适应能力而表现出低多样性高富集的特征。实时荧光定量分析结果也证明, 土壤活性微生物数量随着土壤水分含量的增加而增加, 进而表现出高富集的特征。
土壤水分改变了土壤微生物群落结构, 塑造了不同水分状况下微生物分布特征(图 2)。不同水分条件下东北水稻土优势的微生物门为变形菌门、厚壁菌门以及绿湾菌门。在高水分条件下, 土壤变形菌门相对丰度在土壤总微生物水平和土壤活性微生物水平均显著降低, 具有较差的高水分环境适应能力, 这与周晓丽等的研究结果类似[7]。而厚壁菌门和绿湾菌门在高水分环境下微生物丰度的变化规律与变形菌门相反, 其相对丰度在土壤总微生物水平和土壤活性微生物水平均显著增加, 表明高水分环境刺激了厚壁菌门和绿湾菌门的生长, 进而在高水分环境中显著富集。在精细化分类微生物属水平, 东北水稻土优势的微生物属主要为假单胞菌, 且高水分环境导致土壤总微生物中假单胞菌的丰度显著降低, 而活性微生物中假单胞菌丰度呈增长趋势, 这可能是由于高水分环境下土壤总微生物多样性的增加会导致假单胞菌的丰度降低, 而在土壤活性微生物中, 假单胞菌为关键功能类群, 可能介导了土壤碳氮转化的功能过程。值得注意的是, 在湿润条件下土壤总微生物中假单胞菌占比为38.4%, 但在土壤活性微生物中占比仅占6.7%。类似地, 淹水条件下土壤总微生物中假单胞菌占比31.9%, 而在土壤活性微生物中仅占11.6%。一方面是由于假单胞菌增殖代谢较快, 土壤中残留较多的假单胞菌残体DNA被提取出来, 从而增加了其在土壤总微生物类群中的占比[32];另一方面, 由于假单胞菌细胞较小, 难以通过常规的涡旋方法使其与土壤颗粒分离, 极可能导致大部分假单胞菌通过离心而脱离细胞层, 从而大大降低了假单胞菌的提取效率[19, 33]。土壤中仅有少量的微生物具有生理活性, 研究结果发现, 土壤总微生物细菌数量约为土壤活性微生物细菌数量的70倍。这与Blagodatskaya等[17]的研究结果一致。因此, 深度挖掘这些关键微生物的功能活性, 可为黑土定向培肥提供支撑。
尽管常规提取土壤总微生物与土壤细胞提取方法分辨率不同, 但均能较好地表征土壤中微生物丰度占优势的那部分活性微生物类群(图 3)。例如, 在土壤总微生物水平, 假单胞菌在湿润和淹水条件下均显著增长, 其占比由对照处理的0.2%显著增加到湿润处理的38.4%和淹水处理的31.9%。类似地, 基于土壤活性微生物分析, 湿润和淹水处理假单胞菌的丰度分别占总活性微生物的6.7%和11.6%。在不同水分条件下土壤微生物中的优势类群均为假单胞菌, 并且这一规律在土壤总微生物及活性微生物水平一致。因此, 基于土壤总微生物和活性微生物的高通量测序结果基本反映了关键类群假单胞菌在土壤元素循环过程中的贡献及响应规律。类似地, 针对假单胞菌为核心的微生物类群进行共存微生物网络分析也发现, 高水分条件会使土壤微生物以及假单胞菌共存网络更加松散, 这与之前高水分环境会降低土壤活性微生物多样性的结果一致(图 4)。进一步基于土壤总微生物和活性微生物功能比较分析发现, 尽管不同微生物功能过程在土壤总微生物与土壤活性微生物的占比具有差异, 但其在两种土壤水分状况下的变化趋势较为一致。例如, 化能异养和好氧化能异养过程在土壤元素循环占比高达30%—40%, 但其受水分影响波动较小, 其在土壤总微生物与土壤活性微生物水平表现一致。吴希慧等[34]的研究结果也发现不同土地利用方式对土壤微生物功能的影响主要为化能异养碳转化功能过程。而参与土壤氮循环的功能过程以固氮过程为主, 同时还存在硝酸盐转化过程, 其在土壤总微生物水平和土壤活性微生物水平对水分的响应也较为一致。此外, 实时荧光定量也得到类似的分析结果, 土壤总微生物数量和土壤活性微生物数量均随着土壤水分含量的增加而增加。常规提取土壤总微生物与土壤细胞提取方法均能较好的表征土壤微生物功能类群和功能过程。必须强调的是, 本研究提取土壤细胞DNA所得到的活性微生物群落似乎并不能完全代表土壤中全部的活性微生物组成[35]。一方面由于细胞形态、活性及代谢方式均会影响土壤微生物细胞提取, 进而影响微生物类群丰度。一般而言, 细菌细胞直径范围跨度可达4个数量级, 而古菌细胞更小, 比细菌细胞差异可达10倍之多, 这种差异会改变土壤微生物细胞提取效率[36]。而不同的微生物类群的细胞与土壤颗粒的粘附程度也不相同, 最终也会影响土壤细胞提取效率。另一方面, 不同类型母质土壤的矿物组成显著不同, 也会显著影响土壤微生物组成和提取效率[37]。而且, 本研究仅针对一种水稻土开展研究, 研究结果对旱地、湿地等不同类型土壤微生物类群的普适性仍待进一步验证。
4 结论综上所述, 水分增加显著降低了土壤微生物呼吸活性, 改变了土壤活性微生物群落组成和多样性分布特征。通过提取土壤总DNA和细胞DNA进行高通量测序进一步表征了土壤总微生物和活性微生物类群。淹水处理下土壤活性微生物多样性显著高于湿润处理, 而土壤总微生物多样性却显著降低, 这种差异是由微生物适应能力和微生物间竞争作用共同决定。在土壤总微生物和活性微生物水平, 水分增加均显著增加了土壤微生物数量。微生物共存网络结果表明, 水分增加改变了土壤微生物间的共存模式。与湿润处理相比, 淹水处理显著提升了土壤微生物间的负相关关系, 强化了微生物间的竞争关系。土壤DNA和细胞DNA水平的研究结果表明, 两种水分状况下土壤优势的微生物门均为变形菌门和厚壁菌门, 而优势的属主要为假单胞菌。尽管两种水平在微生物组研究方面的分辨率具有差异, 但是均能较好表征土壤中微生物丰度占优势的关键功能微生物类群。因此, 未来土壤微生物组研究应更加重视科学问题本身对技术手段的内在需求。总而言之, 我们的研究强调了土壤水分对改变土壤活性微生物区系特征和共存模式的重要意义, 研究结果为未来农田土壤微生物组研究提供了新的思路。
| [1] |
梁爱珍, 李禄军, 祝惠. 科技创新推进黑土地保护与利用, 齐力维护国家粮食安全——用好养好黑土地的对策建议. 中国科学院院刊, 2021, 36(5): 557-564. |
| [2] |
高威, 王连峰, 贾仲君. 长期不同施肥模式对农田黑土微生物群落构建的影响. 生态与农村环境学报, 2021, 37(11): 1437-1448. |
| [3] |
Banerjee S K, Helgason B, Wang L F, Winsley T, Ferrari B, Siciliano S. Legacy effects of soil moisture on microbial community structure and N2O emissions. Soil Biology and Biochemistry, 2016, 95: 40-50. DOI:10.1016/j.soilbio.2015.12.004 |
| [4] |
Baveye P. Ecosystem-scale modelling of soil carbon dynamics: Time for a radical shift of perspective?. Soil Biology and Biochemistry, 2023, 184: 109112. DOI:10.1016/j.soilbio.2023.109112 |
| [5] |
杨阳, 刘良旭, 张萍萍, 吴凡, 周媛媛, 宋怡, 王云强, 安韶山. 黄土高原土壤水分-有机碳-微生物耦合作用研究进展. 生态学报, 2023, 43(4): 1714-1725. |
| [6] |
Birch H F. The effect of soil drying on humus decomposition and nitrogen availability. Plant and Soil, 1958, 10(1): 9-31. DOI:10.1007/BF01343734 |
| [7] |
周晓丽, 包丽君, 柳旭, 熊艺, 褚海燕, 贾仲君. 干湿交替水分胁迫对水稻土细菌群落影响的研究. 微生物学报, 2022, 62(3): 1004-1019. |
| [8] |
Sokol N W, Slessarev E, Marschmann G L, Nicolas A, Blazewicz S J, Brodie E L, Firestone M K, Foley M M, Hestrin R, Hungate B A, Koch B J, Stone B W, Sullivan M B, Zablocki O, LLNL Soil Microbiome Consortium, Pett-Ridge J. Life and death in the soil microbiome: how ecological processes influence biogeochemistry. Nature Reviews Microbiology, 2022, 20(7): 415-430. DOI:10.1038/s41579-022-00695-z |
| [9] |
Ma B, Wang H Z, Dsouza M, Lou J, He Y, Dai Z M, Brookes P C, Xu J M, Gilbert J A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in Eastern China. The ISME Journal, 2016, 10(8): 1891-1901. DOI:10.1038/ismej.2015.261 |
| [10] |
Manzoni S, Schimel J P, Porporato A. Responses of soil microbial communities to water stress: Results from a meta-analysis. Ecology, 2012, 93(4): 930-938. DOI:10.1890/11-0026.1 |
| [11] |
Hao D C, Wang L, Gao W, Xie H T, Bao X L, Jia Z J, Wang L F. Disentangling effects of moisture/gas regimes on microbial community, network configuration and nitrogen turnover of black soil. Eurasian Soil Science, 2021, 54(1): S42-S61. |
| [12] |
Marañón-Jiménez S, Asensio D, Sardans J, Zuccarini P, Ogaya R, Mattana S, Peñuelas J. Seasonal drought in mediterranean soils mainly changes microbial C and N contents whereas chronic drought mainly impairs the capacity of microbes to retain P. Soil Biology and Biochemistry, 2022, 165: 108515. DOI:10.1016/j.soilbio.2021.108515 |
| [13] |
Li J Y, Benti G, Wang D, Yang Z L, Xiao R. Effect of alteration in precipitation amount on soil microbial community in a semi-arid grassland. Frontiers in Microbiology, 2022, 13: 842446. DOI:10.3389/fmicb.2022.842446 |
| [14] |
Hartmann M, Six J. Soil structure and microbiome functions in agroecosystems. Nature Reviews Earth & Environment, 2023, 4: 4-18. |
| [15] |
Ochoa-Hueso R, Collins S L, Delgado-Baquerizo M, Hamonts K, Pockman W T, Sinsabaugh R L, Smith M D, Knapp A K, Power S A. Drought consistently alters the composition of soil fungal and bacterial communities in grasslands from two continents. Global Change Biology, 2018, 24(7): 2818-2827. DOI:10.1111/gcb.14113 |
| [16] |
de Vries F T, Griffiths R I, Bailey M, Craig H, Girlanda M, Gweon H S, Hallin S, Kaisermann A, Keith A M, Kretzschmar M, Lemanceau P, Lumini E, Mason K E, Oliver A, Ostle N, Prosser J I, Thion C, Thomson B, Bardgett R D. Soil bacterial networks are less stable under drought than fungal networks. Nature Communications, 2018, 9: 3033. DOI:10.1038/s41467-018-05516-7 |
| [17] |
Blagodatskaya E, Kuzyakov Y. Active microorganisms in soil: critical review of estimation criteria and approaches. Soil Biology and Biochemistry, 2013, 67: 192-211. DOI:10.1016/j.soilbio.2013.08.024 |
| [18] |
文昌丽, 曹伟伟, 唐雪莲, 赵雯淑, 贾仲君, 孟磊. 基于高通量测序的连续传代富集土壤可培养菌菌群变化规律研究. 生态与农村环境学报, 2023, 39(1): 123-135. |
| [19] |
贾仲君, 蔡元锋, 贠娟莉, 杜文斌. 单细胞、显微计数和高通量测序典型水稻土微生物组的技术比较. 微生物学报, 2017, 57(6): 899-919. |
| [20] |
郑燕, 贾仲君. 新一代高通量测序与稳定性同位素示踪DNA/RNA技术研究稻田红壤甲烷氧化的微生物过程. 微生物学报, 2013, 53(2): 173-184. |
| [21] |
鲍士旦. 土壤农化分析 (3版). 北京: 中国农业出版社, 2000.
|
| [22] |
Stubner S. Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreen detection. Journal of Microbiological Methods, 2002, 50(2): 155-164. DOI:10.1016/S0167-7012(02)00024-6 |
| [23] |
Gao W, Xu J S, Zhao J, Zhang H M, Ni Y Y, Zhao B Z, Tebbe C C, Zhang J B, Jia Z J. Prokaryotic community assembly after 40 years of soda solonetz restoration by natural grassland and reclaimed farmland. European Journal of Soil Biology, 2020, 100: 103213. |
| [24] |
Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Peña A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Tumbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QⅡME allows analysis of high-throughput community sequencing data. Nature Methods, 2010, 7: 335-336. |
| [25] |
Louca S, Parfr ey L W, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science, 2016, 353(6305): 1272-1277. |
| [26] |
张君红, 王健宇, 孟泽昕, 何佳, 董政宏, 刘凯茜, 陈文青. 土壤微生物多样性通过共现网络复杂性表征高寒草甸生态系统多功能性. 生态学报, 2022, 42(7): 2542-2558. |
| [27] |
Sollins P, Homann P, Caldwell B A. Stabilization and destabilization of soil organic matter: mechanisms and controls. Geoderma, 1996, 74(1): 65-105. |
| [28] |
Schlüter S, Leuther F, Albrecht L, Hoeschen C, Kilian R, Surey R, Mikutta R, Kaiser K, Mueller C W, Vogel H J. Microscale carbon distribution around pores and particulate organic matter varies with soil moisture regime. Nature Communications, 2022, 13: 2098. |
| [29] |
孙中林, 吴金水, 葛体达, 唐国勇, 童成立. 土壤质地和水分对水稻土有机碳矿化的影响. 环境科学, 2009, 30(1): 214-220. |
| [30] |
Barnard R L, Osborne C A, Firestone M K. Changing precipitation pattern alters soil microbial community response to wet-up under a Mediterranean-type climate. The ISME Journal, 2015, 9(4): 946-957. |
| [31] |
张彬, 刘满强, 钱刘兵, 梁山峰. 土壤微生物群落抵抗力和恢复力: 进展与展望. 生态学报, 2023, 43(14): 5674-5685. |
| [32] |
Warren C R, Manzoni S. When dry soil is re-wet, trehalose is respired instead of supporting microbial growth. Soil Biology and Biochemistry, 2023, 184: 109121. |
| [33] |
Carini P, Marsden P J, Leff J W, Morgan E E, Strickland M S, Fierer N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nature Microbiology, 2016, 2: 16242. |
| [34] |
Lewis W H, Tahon G, Geesink P, Sousa D Z, Ettema T J G. Innovations to culturing the uncultured microbial majority. Nature Reviews Microbiology, 2021, 19: 225-240. |
| [35] |
吴希慧, 王蕊, 高长青, 高胜, 杜兰兰, KHAN Asif, BARMON Million, 郭胜利. 土地利用驱动的土壤性状变化影响微生物群落结构和功能. 生态学报, 2021, 41(20): 7989-8002. |
| [36] |
Sun D L, Jiang X, Wu Q L, Zhou N Y. Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Applied and Environmental Microbiology, 2013, 79(19): 5962-5969. |
| [37] |
Lombard N, Prestat E, van Elsas J D, Simonet P. Soil-specific limitations for access and analysis of soil microbial communities by metagenomics. FEMS Microbiology Ecology, 2011, 78(1): 31-49. |