 2016, Vol. 36
2016, Vol. 36文章信息
- 韩丽丽, 贺纪正
- HAN Lili, HE Jizheng.
- 病毒生态学研究进展
- Advances in viral ecology research
- 生态学报[J]. 2016, 36(16): 4988-4996
- Acta Ecologica Sinica[J]. 2016, 36(16): 4988-4996
- http://dx.doi.org/10.5846/stxb201501210170
-
文章历史
- 收稿日期: 2015-01-21
- 网络出版日期: 2015-12-01

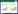




病毒是目前所知的最简单的生命单元,通常是由外壳蛋白和包被在外壳蛋白内的核酸(DNA或RNA)两部分组成。病毒本身缺乏完整的酶系统及能量转化系统,当游离于环境中时,它只是一个有机大分子,只有侵染宿主后才具有生命特征,进行复制。它是地球上最丰富的生物实体,在环境中数量巨大且无处不在,据估计仅海洋中病毒的数量就超过1030。随着现代分子生物学的发展,病毒及其在环境中的作用逐渐被人们所认知,它诱导的细胞裂解间接地促进了环境中有机和无机营养元素的循环,是生物地球化学循环的重要驱动者之一;它能侵染多个宿主,是物种间基因水平转移的重要媒介;它直接推动了微生物物种形成及进化发展,是微生物多样性的关键影响因子;病毒的丰度、多样性及其分布构成了生态系统功能的重要参数。
本文将从病毒的结构与分类、病毒生态学研究方法、病毒的生态功能及土壤病毒生态学研究等方面对病毒生态学的研究进展进行综述。
1 病毒的分类及系统发育1977年,Woese通过比较多种生物的16S rDNA或18S rDNA的核苷酸序列,提出了生物界的三域学说(Three Domains Theory),即细菌域(Bacteria)、古菌域(Archaea)及真核生物域(Eukarya)[1],这三域构成了生物界的生命之树。在该学说中,病毒作为一个重要的生物实体,因其自身不具备繁殖的能力,它的分类地位是游离于这三域之外的。研究发现,生物界中每个生物体通常都被一种或多种病毒所侵染,可以说这三域之间正是通过病毒联系起来的,无论是细菌、古菌还是真核生物,它们的进化过程都和病毒密不可分。
1.1 病毒的结构与分类病毒不同于一般的细胞生物,没有完整的细胞结构,通常由蛋白质外壳包裹着单一核酸(DNA/RNA)组成;它是一种独特的侵染因子,缺乏自身的核糖体,不能进行独立的代谢活动,但其所含的核酸能使病毒利用寄主细胞进行复制。因此,病毒既可被看作是极其简单的微生物,也可被看作是极其复杂的化学物质。图 1是几种常见的病毒类型[1-2],其中有尾噬菌体主要侵染细菌,而球状的包膜病毒主要侵染动物和植物细胞,丝状病毒在环境中分布较少,但它可侵染所有形式的生命细胞。另外环境中还存在一类不具有完整病毒结构的亚病毒(subviruses),包括类病毒、拟病毒、朊病毒。这类简单病毒仅含有某种核酸而不具备蛋白质结构,或仅具有蛋白质而不含核酸,它能够侵染动植物的微小病原体。这里以大肠杆菌病毒T4噬菌体为例,简要介绍一下有尾噬菌体的形态结构及侵染方式。T4噬菌体头部大小为95 nm×65 nm,内部包含了1.66×106个核苷酸的双链线性DNA,尾部是一个100 nm×20 nm的管状器官,由一个内径为2.5 nm的中空尾髓及外面包着的尾鞘组成。尾部末端还有基板、6根尾钉和6根尾丝。在头部和尾部连接处是一个颈圈结构,颈圈上有6根颈须。T4噬菌体通过尾丝和寄主细胞的特异受体结合,通过尾部释放的溶菌酶和尾鞘收缩作用,使尾髓穿透细胞壁和膜,将头部的核酸通过尾髓注入寄主细胞,蛋白外壳则留在细胞外;噬菌体的核酸在细菌寄主体内复制,并与新合成的蛋白质外壳装配成成熟的噬菌体颗粒释放到环境中。

目前,国际上对病毒研究还没有通用的系统发育学分类方法,主要依据病毒宿主、基因组类型、复制方式、基因含量以及形状等方式进行分类。2013年国际病毒分类委员会(International Committee on Taxonomy of Viruses,ICTV)公布的ICTV分类法中,将病毒分为7个目(Orders):有尾噬菌体目(Caudovirales)、疱疹病毒目(Herpesvirales)、线状病毒目(Ligamenvirales)、单股反链病毒目(mononegavirales)、网巢病毒目(Nidovirales)、小核糖核酸病毒(Picornavirales)和芜菁黄花叶病毒目(Tymovirales)。
1.2 噬菌体的分类及系统发育研究噬菌体是一类感染细菌、真菌、放线菌或螺旋体等微生物的病毒的总称,最早是在1915年由英国细菌学家托特发现,至今已经有100a的研究历史[4]。根据噬菌体与被感染寄主菌的相互关系,可将噬菌体分为两大类——烈性噬菌体(virulent phage)和温和噬菌体(temperate phage)或溶源噬菌体(lysogenic phage)。
由于官方的病毒分类信息主要基于病毒表型特征及寄主信息,这对于生态学的研究显然是不够的。因此,美国学者Rohwer等在ICTV分类系统的基础上,基于167个完整的噬菌体基因组序列,通过生物信息学分析首次提出了噬菌体蛋白组树(Phage Proteomic Tree,PPT)的分类方法[5-6],目前该发育树已经囊括了1220个细菌和古菌噬菌体的DNA/RNA基因组序列,具体分类及基因组信息见表 1[5, 7]。PPT系统发育树总体结构与ICTV分类一致,但对于一些遗传信息发生改变,如发生基因水平转移的噬菌体,在PPT分类法中它们的分类地位被真实地还原。如P4噬菌体在ICTV分类法中被归入肌尾病毒科(Myoviridae),但其蛋白组学信息显示其属于短尾病毒(Podophage),可能是因为在进化过程中,它获得了肌尾病毒某个病毒粒子的表型基因,使其表现出了肌尾病毒的形态。
| 噬菌体类群 Phage group | 基因组类型 Genome type | 代表性噬菌体 Typical phage | 基因组大小 (kb) Genome size range (kb) | 基因数量 Quantity of genes |
| 光滑噬菌体 Leviphage | ssRNA 线性 | MS2-like Qβ-like | 3.4—4.3 | 4 |
| 囊状噬菌体 Cystophage | dsRNA 线性 | Φ6-like | 13.3 | 12 |
| 丝状噬菌体 Inophage | ssDNA 环状 | f1-like | 6.0—9.0 | 4—10 |
| 微小噬菌体 Microphage | ssDNA 线性 | ΦΧ174-like Chp1-like | 5.3—6.1 | 10 |
| 短尾噬菌体 Podophage | dsDNA 线性 | Φ29-like T7-like Mx8-like N4-like P1-like P22-like P4-like | 16.0—70.0 | 20—65 |
| 长尾噬菌体 Siphophage | dsDNA 线性 | λ-like D29-like T5-like A118-like Mu-like SPP1-like T1-like TPA2-like TP901-1-like | 22.0—121.0 | 36—88 |
| 肌尾噬菌体 Myophage | dsDNA 线性 | T4-like P2-like ΦCbK-like Pr-like SN-like | 33.0—244.0 | 40—415 |
| Fuselloviridi | dsDNA 环状 | SSV—1 | 14.8—17.3 | 31—37 |
噬菌体在环境中具有两个重要功能,一是作为病原体侵染微生物,改变环境中微生物的数量、群落结构以及功能。由于噬菌体对寄主具有特异性且侵染过程通常发生在寄主密度较高的情况下,即高接触率可导致高侵染率,因此关于噬菌体在环境中的侵染机制,有人提出了杀死优胜者假说。关于这一假说目前学术界一直争论不休,Diaz-Munoz在应用微生物学研究进展一书中罗列了大量关于论证或驳斥该假说的文献[8]。总的来讲,噬菌体对微生物群落的影响也要视环境因子而定,同样的环境中噬菌体可能对某些细菌种有影响而对群落中的其它种类细菌没有影响。另外,牛津大学的研究人员Vos等发现,土壤中的噬菌体能够根据其周围环境中细菌的类型而不断改进它们感染细菌的能力[9],说明细菌不断进化的抗侵染能力缓冲了一部分噬菌体侵染的效果,这种进化方式有利于重构其生存的小范围生态系统。
噬菌体在环境中的另一个重要功能是作为微生物间的穿梭载体,促进细菌间基因的横向转移,使其获得更为有利的基因快速适应环境的变化。噬菌体侵染细菌和细菌抵抗侵染的机制是细菌进化的关键,它涉及到噬菌体的功能吸附[10]、寄主及噬菌体的结构变化[11],噬菌体核酸向细菌细胞转运并避免核酸在细胞内降解[12],最终导致噬菌体和细菌表型的变化等几个过程。病毒还能交换自身和寄主的基因信息,该作用机制可能在进化过程中直接推动了原核生物物种的形成[13]。另外,有研究指出寄主对噬菌体的敏感性或抗性取决于寄主所含的质粒,噬菌体侵染促使核酸和寄主质粒整合后,质粒在不同的细菌之间转移,可导致噬菌体寄主谱系变宽[12, 14]。随着研究的深入,越来越多的证据显示,无论是在自然条件下还是实验室环境中,细菌与噬菌体的这种共进化关系能增加它们的表型及遗传多样性,增强细菌和噬菌体的进化及分异速率,推动微生物群落的演变。
另外,噬菌体还能影响寄主的生理活性,如通过编码寄主的同源基因影响寄主菌的代谢,在某些情况下,这种辅助的代谢基因可被用来增强寄主细胞的生理活性。如蓝藻噬菌体所携带的光合基因可帮助寄主细胞在光合蛋白消失后为其提供能量[15]。很多蓝藻噬菌体侵染蓝藻后,会在宿主体内表达编码光合作用Ⅱ反应中的核心蛋白,如psbA基因编码的D1蛋白,它能在所有产氧光合作用生物间快速的转移光合作用产物。而病毒的复制速率则取决于光合作用速率,因此侵染过程中这些光合作用基因的表达不仅增强宿主的光合作用速率,也增加蓝藻噬菌体的复制速率。
2 病毒生态学研究方法过去人们对自然界中存在的病毒的认识主要是依靠显微镜观察和分离培养。在研究病毒生态学过程中,研究者们需要突破两大难点,一是大部分的噬菌体宿主目前不可培养,二是对病毒的研究缺乏通用的标记基因,因此在研究过程中需要用到一种或多种分析方法。
在20世纪,科学家们对病毒的研究主要依赖于电镜技术研究可培养病毒形态及多样性,这种方法相对便宜且便于操作,缺点是不能准确地评价病毒群体的多样性。随着技术的发展,分子生物学技术被逐渐应用于病毒生态学的研究,最常用的有PFGE(Pulse Field Gel Electrophoresis)和RAPD(Random Amplified Polymorphic DNA analysis)这两种指纹图谱技术,这类技术可用于大量样品的比较分析,相对经济、易操作,还可发现新型病毒,缺点是同显微技术一样,不能评价病毒群体的多样性,也不能提供直接的功能信息。由于病毒的研究缺乏通用的标记基因,因此很多科学家也选择针对某一类病毒,以一些高度保守的结构或功能基因作为标记基因,研究病毒的系统发育地位,常见的基因有T4噬菌体的g23基因,蓝藻噬菌体的g20、psbA和psbD基因等。
进入21世纪,高通量测序技术的出现,突破了探寻环境微生物黑箱的瓶颈。关于病毒基因组的测序始于1977年,第一株被测序的病毒是侵染大肠杆菌的ΦΧ174噬菌体,基因组共包含5 375个核苷酸[16]。2002年,Breitbart等首次通过鸟枪测序法对两个海洋病毒群体进行了宏基因组测序,初探了海洋环境中病毒的多样性,结果表明这两个海水样本中病毒多样性极高,有超过65%的序列之前没有被报道过[17]。2005年,Edwards等提出了病毒宏基因组的概念,并针对环境中的噬菌体群体提出了新的PPT分类方法[6]。西班牙的Rodriguez-Valera教授实验室利用宏基因组测序技术,测定了超过1000种海洋病毒的遗传序列,发现了208种为新的海洋噬菌体[18]。Hurwitz等采用比较基因组学和网络分析方法分析了海洋病毒群体的生态驱动模型[19]。经过10余年的研究,病毒宏基因组学技术的发展已经帮助人们从多样的环境中快速有效地发现了许多未知的新病毒和基因。
3 病毒的生态功能 3.1 病毒的传播与应用提到病毒,人们脑海中马上就会浮现出艾滋病毒、SARS病毒、禽流感病毒以及近期在西非蔓延的埃博拉病毒等,这些病毒严重威胁人类的健康和生命安全。自古以来,人类就在和病毒进行不断的斗争,如古代的天花、牛痘等,病毒威胁着人类生存的同时也促进人类不断完善自身的免疫防御系统。病毒除了威胁人类健康,也会侵害植物,其中我国的水稻、小麦、玉米等农作物病毒病害发生严重,给社会造成了重大的经济损失。作物病害中常见的病毒有水稻条纹病毒、水稻黑条矮缩病毒、小麦黄花叶病毒、玉米粗缩病毒、烟草花叶病毒等等。植物病毒不像噬菌体和动物病毒那样具有壳体蛋白与寄主细胞表面受体之间的特异性,一般在植物细胞表面损伤后,从伤口侵入。自然界中植物病毒的传播主要依靠昆虫作为媒介(如蚜虫、叶蝉等)。了解病毒在环境中的类型、丰度、生存和分布状态,有助于人们对环境生态系统健康进行评价。
凡事都有两面性,病毒除了能使人和动植物致病外,也有对人类有利的一面。例如针对某些特殊病毒的增殖、失活、存活、以及生命周期等特性,调控感染农业有益(根瘤菌)或有害微生物(植物病原体)的病毒使其朝着对作物生产有利的方向发展。自1915年噬菌体被发现以来,土壤生物学家在农业生产方面开展了很多关于根瘤菌噬菌体(rhizobiphage)的研究。包括种群、根瘤菌噬菌体宿主范围、根瘤菌噬菌体对根瘤形成的影响、根瘤菌噬菌体对豆科植物产量等的研究[20],研究还发现种植豆科植物的土壤中存在的大量根瘤菌噬菌体丰度和宿主细菌的丰度趋势一致[21]。另外,科学家们还针对病毒对土传植物病害的影响进行研究,例如Ashelford等人在甜菜地中观察到了短尾噬菌体(Podoviridae)及其宿主液化沙雷氏菌(Serratia liquefaciens)在土壤中均存在显著的季节性种群动态变化特征[22]。
从医学和人类健康角度来讲,病毒侵染病原细菌,如霍乱弧菌噬菌体、炭疽芽孢菌噬菌体等能将病原菌宿主裂解,可作为治疗这些疾病的特效药,从而减少流行病的发生,这种通过噬菌体裂解细菌治疗病原菌感染的手段称为噬菌体疗法(Phage Therapy)。噬菌体疗法最早在1919年被应用于4名法国儿童痢疾的治疗,结果这4名儿童均恢复了健康[23]。20世纪中期,由于抗生素的发展大行其道,欧美等国家对噬菌体的研究一度处于停滞阶段。随着病原菌对抗生素抗性问题日益严重,导致了大量病原菌产生耐药性,甚至出现超感染免疫抗性,正是病原菌对抗生素的这种耐药性促进了噬菌体研究的复兴。2014年5月召开的美国微生物学会(ASM)上,瑞士洛桑大学的Resch博士首次介绍了由欧盟投资380万欧元的Phagoburn计划,这是首个应用噬菌体疗法治疗人体感染的大型跨国临床试验。因噬菌体的宿主谱系较窄,只针对特异的寄主病原菌起作用,特异性高、对人体细胞无害等特点,较抗生素这类广谱杀菌剂而言有一定优势,科学家们正在研究通过噬菌体疗法代替抗生素达到控制病原菌的目的,并将该技术逐步应用于医学、食品、水产、农业、环境等多个领域[24]。
3.2 病毒与元素的生物地球化学循环病毒还是生物地球化学循环的重要驱动者。在海洋环境中,病毒介导的细胞裂解所释放的碳源和其他营养元素推测是全球范围碳循环的主要驱动因子[25]。地球早期的生命形态大部分都是化能自养型(chemolithoautotrophs),它们所需的能量主要来自周围的岩石和海洋;细菌细胞组分可直接固定环境中的CO2,这种不产氧光合作用在早期的地球上很丰富,只通过光照和CO2产生它们的生命形式。而如今细菌中所参与的各种各样的代谢活性,有许多和人类生理代谢很相似,比如能使人致病的葡萄球菌(Staphylococcus)、沙门氏菌(Salmonella),或者食品和饮料如奶酪和酒中的异养生物(heterotrophs),利用复杂的碳水化合物或其它有机分子作为细胞能量,组成细胞组份,这些都不同于早期的岩石和光照作为能量来源[26]。那么早期的这些异养生物是如何开发利用环境中庞大的有机碳资源的呢?推测可能是病毒侵染导致这些细菌和古菌的细胞凋亡,释放大量的有机碳进入海洋,为异养生物开拓了新的资源利用领域,同时也将新的碳固定环节带入碳循环。
地球化学研究证据指出地球大气中直到40亿年前才出现氧气分子,随后游离氧浓度逐步增加至今天的21%,这个过程称为大氧化事件[27]。早期研究生物驱动大气成份的改变主要是通过蓝藻,蓝藻也是现今开展这类研究最常用的模式生物之一,全球海洋中关于光合作用的研究大部分是由原绿球藻(Prochlorococcus)和聚球藻(Synechococcus)这两种最常见的蓝藻驱动的,它们贡献了全球碳固定量的25%[28]。蓝藻光合作用过程中的部分活性还要归功于它的合作伙伴——蓝藻噬菌体。蓝藻噬菌体和蓝藻光合作用能力的协同进化是产氧光合作用的远古驱动者,也是空气中氧气含量在地球表面升高的早期驱动者。有研究指出地球上每20个人中就有1个人呼吸的氧气是来自海洋中病毒刺激的生氧光合作用。
当病毒裂解其微生物宿主时,会导致惊人数量的生源要素释放到环境中去。在海洋中,病毒甚至会重复利用这些被特定的海洋大型浮游生物所消耗的营养物质。病毒裂解沿海水域“褐潮”中的藻类Aureococcus anophagefferns,还可增加海水中溶解的铁含量,随后溶解的铁又被异养细菌快速转移为可过滤的颗粒物[29]。此外,病毒甚至还能影响到天气,如病毒侵染藻类时会释放二甲基硫(DMS),DMS 不仅与酸雨、酸雾的形成有关,它还能使大气中的云凝结核,增加对太阳的辐射[30]。
据估计地球上原核生物的数量在1030[31],而病毒丰度约为1031[32];海洋环境学家们认为海洋中病毒颗粒至少是原核生物的10倍,这还不包括整合在基因组上的溶源性病毒,且病毒在环境中每秒可发生高达1024次的寄主侵染,以维持它们在环境中的数量[7],这些数据间接证明了病毒对生物地球化学循环起着关键作用,估算土壤中微生物和噬菌体的丰度对评估土壤中病毒在元素的生物地球化学循环方面有着重要作用。目前,噬菌体在环境中的这些特征和数据主要是通过海洋中噬菌体的研究所得,而土壤中的噬菌体在环境条件发生改变时,它们的群体结构会作出更加迅速的反应[33],因此可以推测噬菌体在土壤中发挥着更为重要的作用。
3.3 量化病毒对生态系统的贡献需要技术及数据分析方法的进步病毒、细菌以及其他生物在环境中的相互关系是相对稳定的。在海洋环境中,病毒贡献了很多和能量代谢相关的基因。研究者们通过显微镜观察、关键代谢产物产生速率及决定性的病毒作用过程速率粗略的评估病毒对整个能量流的贡献,但精确的值还是很难获得。更为复杂的系统,如肠道和土壤,有数千种病毒类型存在,由于病毒间缺乏通用的标记基因,加上病毒变异速率快,功能基因保守性低等特性,目前还无法通过分子生物学方法对病毒数量进行量化,评估复杂环境中病毒活性则更为困难。目前国际上已经有不少实验室的科学家们正专注这一新领域的研究并尝试进一步了解全球生态系统对病毒的依赖,如开发生物反应器等人工模拟系统,这将有利于人们进一步认识病毒如何驱动生产力的发展并维持生态系统的稳定。
4 土壤病毒生态学研究进展过去几十年由于病毒生态学的迅速发展,被称为病毒学研究的第三纪[34],但这个阶段的研究主要集中在海洋和湖泊等水生环境,在土壤环境中涉及的还很少。土壤中的病毒生态学的研究起步于近10年,且集中在湿地和稻田等接近水生生态系统的噬菌体研究。如借助对海洋病毒的认知,利用已知海洋病毒的基因组序列设计蓝藻噬菌体及T4噬菌体功能基因的引物,考察这两种病毒在土壤中的多样性及分布特征。国内的王光华等和日本的Kimura教授合作在蓝藻噬菌体和T4噬菌体的多样性研究方面发表了多篇论文。他们通过蓝藻噬菌体的g20基因研究了稻田田面水和稻田土壤中噬藻体的多样性[35],同时比较了日本水稻土和中国东北水稻土中蓝藻噬菌体的g20基因序列,发现两个地区的蓝藻噬菌体分布存在明显差异。以g23基因作为标记基因研究了水稻土[36-37]、湿地[38]及旱地[39]黑土中T4噬菌体的多样性,研究表明g23基因分布与其生存环境关系很大,T4 型噬菌体基因多样性非常丰富,它的群落结构在湖泊、海洋和稻田环境中具有较大的差异。由于研究中所使用的g20和g23基因的扩增引物均依据海洋分离的噬菌体序列设计,加之噬菌体本身快速突变的特性,这两个基因扩增所得序列专一性并不高,目前只能通过变性梯度凝胶电泳(DGGE)及切胶测序的方法分析这两种噬菌体的多样性,还未见利用高通量测序方法研究这两种噬菌体多样性的报道。
环境中病毒的群体组成是复杂而多样的,除了对土壤中T4和蓝藻噬菌体等单一病毒多样性及分布特征研究外,搞清楚土壤中有哪些病毒,这些病毒的分布和土壤环境因子之间的关系如何也很重要。早期有一些基于表型的研究表明病毒的多样性与土壤的基本性质密切相关。如病毒在低温时潜伏期变长、生活周期也变长,噬菌体的释放量急剧降低,推测是由于温度较高的情况下,微生物和酶的活性更高,蛋白酶等加速了病毒的分解代谢周期[20]。可见同细菌相似,温度也是病毒在土壤中生存的重要控制因子。Williamson等通过对6种美国东部地区的土壤(2种农田土壤,2种沿海平原森林土壤和2种典型山脉森林土壤)中的噬菌体多样性研究表明森林土壤中噬菌体的含量要高于农田土壤,噬菌体的含量和水份含量相关而和土壤质地关系不大,土地使用类型和细菌及病毒的丰度均显著相关[40],湿润的森林土壤中的病毒丰度和多样性均要高于旱地农田土壤,土地利用方式是影响病毒丰度及多样性的关键因素[33, 40-41]。由此可见,病毒的多样性及其与宿主的相互作用受温度、pH、含水率、土壤结构及营养元素可利用性等多种环境因子的影响。
不得不说近年来,宏基因组学作为环境微生物学的前沿研究方法,为宏病毒组学的发展带来了新的契机。最近Reavy等在AEM上发表了1篇4种不同类型土壤(沿海土、耕作棕壤土、高山灰壤土和腐殖质铁灰壤土)中病毒多样性的文章[42],通过宏基因组学结合表型分类方法比较不同类型土壤中的病毒,研究结果表明这几种土壤中富集的病毒以球状病毒颗粒和细丝状病毒为主;高通量测序结果显示在沿海土壤和耕作土这两种土壤宏病毒组中均以单链环状DNA病毒(ssDNA)占主导地位,其中沿海土中Gokushovirus(侵染衣原体、螺原体等原核寄生生物的病毒)较耕作土明显占优势,并推测ssDNA病毒的分布特征可能由土壤理化因子驱动的。今年Adriaenssens等也发表了1篇基于宏基因组学方法分析纳米布(Namib)沙漠土壤中病毒群体结构的文章[43]。纳米布沙漠位于非洲西南部大西洋沿岸的干燥区,是世界上最古老、最干燥的沙漠之一。文章基于电子显微镜的表型技术以及Illumina平台的宏病毒组高通量测序技术研究发现该沙漠土壤中的病毒主要分布于有尾噬菌体目,尤其以长尾噬菌体为主。对测序所得的功能基因与4个病毒基因数据库(KEGG、COG、SEED和ACLAME)比对后发现细胞壁裂解酶基因、核苷酸还原酶基因以及噬菌体相关基因丰度较高。以焦磷酸水解酶基因phoH和末端酶大亚基基因terL作为标记基因,构建的系统发育树显示纳米布沙漠土壤中的病毒基因大多是新的未报道的基因。可见病毒基因数据库特别是土壤中的病毒基因数据库远没有其它微生物基因信息完善,宏病毒组测序所得序列通常有一半不能和数据库中已知的序列相匹配,这也是制约病毒生态学研究发展的主要因素。随着科技的进步和大数据时代信息的发展,我们相信土壤病毒生态学研究将会有长足的进步和更为广阔的发展空间。
5 展望从1915年英国细菌学家托特发现噬菌体至今,有关微生物病毒的研究已经整整100a,噬菌体的发现和研究彻底改变了我们对生物学的理解。随着研究技术的不断发展,针对环境中病毒的研究经历了最初的显微技术、单个或多个基因的分子生物学技术、单个病毒的基因组学技术以及环境病毒的宏基因组学技术这几个阶段,尤其是高通量技术的应用大大促进了病毒生态学研究的发展。这些技术从不同层面揭示了病毒基因的多样性,展示了病毒在地球生物化学循环中的重要驱动作用。在病毒研究的下一个世纪中,在土壤病毒生态学方向可重点开展以下几方面的研究:(1)从大尺度上研究土壤中病毒的群体组成及空间分布格局,揭示土壤病毒地理分异的生态驱动机制;(2)对病毒裂解宿主菌,导致营养元素的循环进行量化研究,用以评估土壤中的病毒在元素生物地球化学循环过程中的作用;(3)结合各种组学技术对土壤中的病毒进行研究,在表型、基因和功能水平上进一步解析病毒与宿主菌之间的协同进化关系;(4)理解不同类型病毒在土壤环境中的生存和传播机制,为病毒的控制提供依据。
| [1] | Woese C R, Fox G E. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences , 1977, 74 (11) : 5088–5090. DOI:10.1073/pnas.74.11.5088 |
| [2] | Adenosine. File: Tevenphage.svg. Wikimedia. (2008-5-28) [2015-05-13]. http://en.wikipedia.org/wiki/File:Tevenphage.svg. |
| [3] | Rohwer F, Barott K. Viral information. Biology & Philosophy , 2013, 28 (2) : 283–297. |
| [4] | Twort F W. An investigation on the nature of ultra-microscopic viruses. The Lancet , 1915, 186 (4814) : 1241–1243. DOI:10.1016/S0140-6736(01)20383-3 |
| [5] | Rohwer F, Edwards R. The phage proteomic tree: a genome-based taxonomy for phage. Journal of Bacteriology , 2002, 184 (16) : 4529–4935. DOI:10.1128/JB.184.16.4529-4535.2002 |
| [6] | Edwards R A, Rohwer F. Viral metagenomics. Nature Reviews Microbiology , 2005, 3 (6) : 504–510. DOI:10.1038/nrmicro1163 |
| [7] | Rohwer F, Youle M, Maughan H, Hisakawa N. Life in Our Phage World. San Diego: Wholon , 2014 : 8–8. |
| [8] | Díaz-Muñoz S L, Koskella B. Bacteria-phage interactions in natural environments. Advances in Applied Microbiology , 2014, 89 : 135–183. DOI:10.1016/B978-0-12-800259-9.00004-4 |
| [9] | Vos M, Birkett P J, Birch E, Griffiths R I, Buckling A. Local adaptation of bacteriophages to their bacterial hosts in soil. Science , 2009, 325 (5942) : 833. DOI:10.1126/science.1174173 |
| [10] | Randall-Hazelbauer L, Schwartz M. Isolation of the bacteriophage lambda receptor from Escherichia coli. Journal of Bacteriology , 1973, 116 (3) : 1436–1446. |
| [11] | Mahony J, Van Sinderen D. Structural aspects of the interaction of dairy phages with their host bacteria. Viruses , 2012, 4 (9) : 1410–1424. |
| [12] | Richter C, Chang J T, Fineran P C. Function and regulation of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated (Cas) systems. Viruses , 2012, 4 (10) : 2291–2311. |
| [13] | Weinbauer M G, Rassoulzadegan F. Are viruses driving microbial diversification and diversity?. Environmental Microbiology , 2004, 6 (1) : 1–11. |
| [14] | Deng Y M, Harvey M L, Liu C Q, Dunn N W. A novel plasmid-encoded phage abortive infection system from Lactococcus lactis biovar.diacetylactis. FEMS Microbiology Letters , 1997, 146 (1) : 149–154. DOI:10.1111/j.1574-6968.1997.tb10185.x |
| [15] | Mann N H, Cook A, Millard A, Bailey S, Clokie M. Marine ecosystems: bacterial photosynthesis genes in a virus. Nature , 2003, 424 (6950) : 741. |
| [16] | Sanger F, Air G M, Barrell B G, Brown N L, Coulson A R, Fiddes J C, Hutchison C A, Slocombe P M, Smith M. Nucleotide sequence of bacteriophage φX174 DNA. Nature , 1977, 265 (5596) : 687–695. DOI:10.1038/265687a0 |
| [17] | Breitbart M, Salamon P, Andresen B, Mahaffy J M, Segall A M, Mead D, Azam F, Rohwer F. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences of the United States of America , 2002, 99 (22) : 14250–14255. DOI:10.1073/pnas.202488399 |
| [18] | Mizuno C M, Rodriguez-Valera F, Kimes N E, Ghai R. Expanding the marine virosphere using metagenomics. PLoS Genetics , 2013, 9 (12) : e1003987. DOI:10.1371/journal.pgen.1003987 |
| [19] | Hurwitz B L, Westveld A H, Brum J R, Sullivan M B. Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proceedings of the National Academy of Sciences of the United States of America , 2014, 111 (29) : 10714–10719. DOI:10.1073/pnas.1319778111 |
| [20] | Kimura M, Jia Z J, Nakayama N, Asakawa S. Ecology of viruses in soils: past, present and future perspectives. Soil Science and Plant Nutrition , 2008, 54 (1) : 1–32. DOI:10.1111/j.1747-0765.2007.00197.x |
| [21] | Sharma R S, Mohmmed A, Babu C R. Diversity among rhizobiophages from rhizospheres of legumes inhabiting three ecogeographical regions of India. Soil Biology and Biochemistry , 2002, 34 (7) : 965–973. DOI:10.1016/S0038-0717(02)00030-5 |
| [22] | Ashelford K E, Norris S J, Fry J C, Bailey M J, Day M J. Seasonal population dynamics and interactions of competing bacteriophages and their host in the rhizosphere. Applied and Environmental Microbiology , 2000, 66 (10) : 4193–4199. DOI:10.1128/AEM.66.10.4193-4199.2000 |
| [23] | Borysowski J, Międzybrodzki R, Górski A. Phage Therapy. Norfolk: Caister Acadimic Press , 2014 : 289–301. |
| [24] | Jassim S A A, Limoges R G. Natural solution to antibiotic resistance: bacteriophages ‘The Living Drugs’. World Journal of Microbiology and Biotechnology , 2014, 30 (8) : 2153–2170. DOI:10.1007/s11274-014-1655-7 |
| [25] | Jover L F, Effler T C, Buchan A, Wilhelm S W, Weitz J S. The elemental composition of virus particles: implications for marine biogeochemical cycles. Nature Reviews Microbiology , 2014, 12 (7) : 519–528. DOI:10.1038/nrmicro3289 |
| [26] | Greene S E, Reid A. Viruses throughout life & time: friends, foes, change agents. A report on an American academy of microbiology colloquium. San Francisco: American Society for Microbiology , 2013 . |
| [27] | Lyons T W, Reinhard C T, Planavsky N J. The rise of oxygen in Earth's early ocean and atmosphere. Nature , 2014, 506 (7488) : 307–315. DOI:10.1038/nature13068 |
| [28] | Flombaum P, Gallegos J L, Gordillo R A, Rincón J, Zabala L L, Jiao N Z, Karl D M, Li W K W, Lomas M W, Veneziano D, Vera C S, Vrugt J A, Martiny A C. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proceedings of the National Academy of Sciences of the United States of America , 2013, 110 (24) : 9824–9829. DOI:10.1073/pnas.1307701110 |
| [29] | Gobler C J, Hutchins D A, Fisher N S, Cosper E M, Sanudo-Wilhelmy S A. Release and bioavailability of C, N, P, Se, and Fe following viral lysis of a marine chrysophyte. Limnology and Oceanography , 1997, 42 (7) : 1492–1504. DOI:10.4319/lo.1997.42.7.1492 |
| [30] | 王斐, 郑天凌, 洪华生. 海洋病毒在微生物食物环中的重要作用. 海洋科学 , 1998 (4) : 41–43. |
| [31] | Whitman W B, Coleman D C, Wiebe W J. Prokaryotes: the unseen majority. Proceedings of the National Academy of Sciences of the United States of America , 1998, 95 (12) : 6578–6583. DOI:10.1073/pnas.95.12.6578 |
| [32] | Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus?. Trends in Microbiology , 2005, 13 (6) : 278–284. DOI:10.1016/j.tim.2005.04.003 |
| [33] | Srinivasiah S, Bhavsar J, Thapar K, Liles M, Schoenfeld T, Wommack K E. Phages across the biosphere: contrasts of viruses in soil and aquatic environments. Research in Microbiology , 2008, 159 (5) : 349–357. DOI:10.1016/j.resmic.2008.04.010 |
| [34] | Mann N H. The third age of phage. PLoS Biology , 2005, 3 (5) : e182. DOI:10.1371/journal.pbio.0030182 |
| [35] | Jing R Y, Liu J J, Yu Z H, Liu X B, Wang G H. Phylogenetic distribution of the capsid assembly protein gene (g20) of cyanophages in paddy floodwaters in Northeast China. PloS One , 2014, 9 (2) : e88634. DOI:10.1371/journal.pone.0088634 |
| [36] | Wang G H, Murase J, Taki K, Ohashi Y, Yoshikawa N, Asakawa S, Kimura M. Changes in major capsid genes (g23) of T4-type bacteriophages with soil depth in two Japanese rice fields. Biology and Fertility of Soils , 2009, 45 (5) : 521–529. DOI:10.1007/s00374-009-0362-2 |
| [37] | Liu J J, Wang G H, Wang Q, Liu J D, Jin J, Liu X B. Phylogenetic diversity and assemblage of major capsid genes (g23) of T4-type bacteriophages in paddy field soils during rice growth season in Northeast China. Soil Science and Plant Nutrition , 2012, 58 (4) : 435–444. DOI:10.1080/00380768.2012.703610 |
| [38] | Zheng C Y, Wang G H, Liu J J, Song C C, Gao H X, Liu X B. Characterization of the major capsid genes (g23) of T4-type bacteriophages in the wetlands of northeast China. Microbial Ecology , 2013, 65 (3) : 616–625. DOI:10.1007/s00248-012-0158-z |
| [39] | Wang G H, Yu Z H, Liu J J, Jin J, Liu X B, Kimura M. Molecular analysis of the major capsid genes (g23) of T4-type bacteriophages in an upland black soil in Northeast China. Biology and Fertility of Soils , 2011, 47 (3) : 273–282. DOI:10.1007/s00374-010-0533-1 |
| [40] | Williamson K E, Radosevich M, Wommack K E. Abundance and diversity of viruses in six Delaware soils. Applied and Environmental Microbiology , 2005, 71 (6) : 3119–3125. DOI:10.1128/AEM.71.6.3119-3125.2005 |
| [41] | Williamson K E, Wommack K E, Radosevich M. Sampling natural viral communities from soil for culture-independent analyses. Applied and Environmental Microbiology , 2003, 69 (11) : 6628–6633. DOI:10.1128/AEM.69.11.6628-6633.2003 |
| [42] | Reavy B, Swanson M M, Cock P J, Dawson L, Freitag T E, Singh B K, Torrance L, Mushegian A R, Taliansky M. Distinct circular single-stranded DNA viruses exist in different soil types. Applied and Environmental Microbiology , 2015, 81 (12) : 3934–3945. DOI:10.1128/AEM.03878-14 |
| [43] | Adriaenssens E M, Van Zyl L, De Maayer P, Rubagotti E, Rybicki E, Tuffin M, Cowan D A. Metagenomic analysis of the viral community in Namib Desert hypoliths. Environmental Microbiology , 2015, 17 (2) : 480–495. DOI:10.1111/1462-2920.12528 |
{kind=link}